


3. Избыток ФЕН идет на минорный путь метаболизма. Во-первых, сам избыток ФЕН токсичен для организма, а теперь образуются еще более токсичные фенилпируват, фениллактат, фенилацетат.
(На ВН4 не обращайте внимание, это другой тип фенилкетонурии, при нарушении синтеза кофермента)
Делаем вывод, с чем связаны проявления фенилкетонурии: 1) ФЕН токсичен и накапливается, В избытке ФЕН возникает конкурентное ингибирование и все ферменты нормального обмена ФЕН и ТИР ингибируются. 2)ТИР необходимый для синтеза меланинов, дофамина, норадреналина, адреналина не образуется. 3) Минорные пути определяют формирование более токсичных метаболитов.
Сами проявления:
1)Сам ФЕН в избытке токсичен для нейронов – нарушение дифференцировки клеток ЦНС – «фенилаланиновое слабоумие».
2)Метаболиты ФЕН (кетоацидоз): мышиный запах мочи, прямое токсическое действие на ГМ – неврологические симптомы
3)Недостаток ТИР: неврологические нарушение вследствие недостатка нейротрансмиттеров, в т.ч эпилептические припадки и нарушение симпатической иннервации всех внутренних органов (А, НА), гипопигментация (меланины), гипотиреоз, т.е. кретинизм. (Т3,Т4).
13. Молекулярные наследственные болезни углеводного и аминокислотного, белкового обмена. Галактоземия, гликогенозы. Фенилкетонурия, альбинизм.
Молекулярные наследственные болезни = генные болезни.
Практически все наследственные болезни обмена веществ аутосомно-рецессивные.

Галактоземия – наследственное аутосомно-рецессивное моногенное заболевание.
1.Мутация в гене галактозо-1-фосфатуридилтрансферазы
2.Результат: галактоза не превращается в глюкозу, галактоземия (у детей – питание) и снижение глюкозы.
2.1.Галактоза – основной субстрат получения энергии у детей (через превращение в глюкозу). АТФдефицит, т.е. недоразвитие органов и систем в условиях энергодефицита: нарушение дифференцировки клеток мозга – слабоумие; нарушение соотношение тормозных и возбуждающих медиаторов: судороги.
3.Галактоза накапливается и откладывается во внутренних органах (в большей степени паренхиматозных: печень, почки, селезенка, хрусталик, реже – ЦНС, легкие)
3.1.Накопление галактозы в этих органах приводит к формированию дистрофии и функциональной недостаточности, то есть постепенно формируется печеночная, почечная, реже дыхательная недостаточность и энцефалопатия, катаракта (у галактозы тропность к хрусталику).
Гликогенозы – группа аутосомно-рецессивных заболеваний, обусловленных недостаточностью какоголибо фермента, участвующего в синтезе или распаде гликогена.
Здесь проявляется такое свойство мутантного гена, как способность к генокопированию. Генокопия – генетически разнородные заболевания (отсутствуют разные ферменты), имеющие одинаковую клиническую картину.
1.Как минимум 8 мутаций в генах различных ферментов (независимо) приводят к нарушению превращения гликогена в глюкозу, т. е процесса гликогенолиза.
2.Выделяют 3 формы гликогенозов
- Печеночная (гликоген не распадается и накапливается прежде всего в печени). Самое частое I. Болезнь Гирке (дефицит глюкозо-6-фосфатазы)

-Мышечная (гликоген не распадается и накапливается преимущественно в поперечно-полосатых мышцах)
-Смешанная (и в печени и в мышцах, прежде всего в миокарде). Болезнь II. Помпе
3.Проявления.
·Накопление гликогена – дистрофия органов, развитие функциональной недостаточности 1. Печени, 2. Сердца и скелетных мышц, 3. Почек (тоже депо гликогена, но в меньшей степени, чем 1 и 2).
·Нарушение превращения гликогена в глюкозу. Это ощущается в экстремальных условиях, когда требуется больше глюкозы (болезнь, рост, психоэмоциональные нагрузки). Наблюдается гипогликемия, активируется липолиз, жирные кислоты отправляются на синтез кетоновых тел
– кетоацидоз.
Для общей картины. Запоминать все не нужно, только I и II.
Энзимопатии обмена аминокислот

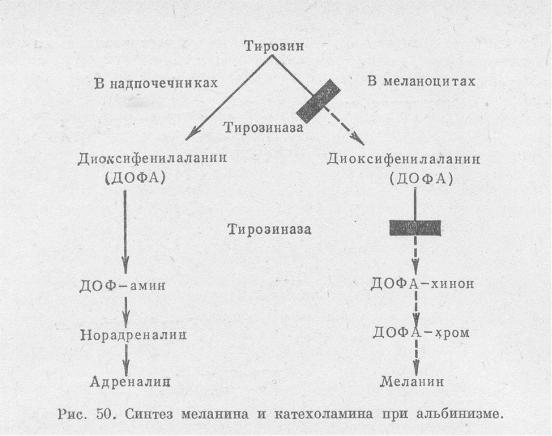
*Про фенилкетонурию смотри в 12 вопросе Альбинизм (полный/глазно-кожный) – аутосомно-рецессивное заболевание, характеризующееся
нарушением синтеза фермента тирозиназы меланоцитов. Синтез катехоламинов при этом не нарушен. *Глазной альбинизм – наследуется по Х-сцепленному рецессивному типу. Проявления со стороны органа зрения.
Проявления:
1.Гипопигментация и сухость кожи, белые пятна на коже. Высокая чувствительность к солнечным лучам – высокая вероятность рака кожи.
2.Нарушение потоотделения
3.Волосы белого или светлого цвета
4.Красная или голубая с розовым оттенком радужка глаз (просвечивают сосуды). Характерна социофобия.
5.Со стороны зрения может быть: косоглазие, амблиопия, нистагм, снижение остроты зрения, функциональная слепота.

14.Хромосомные мутации. Наследственные болезни, обусловленные изменениями в аутосомах
ив половых хромосомах.
Хромосомные мутации характеризуются аномальной структурой отдельных хромосом в связи с изменением их числа или положением генов (первые два столбика соответственно).
Хромосомные болезни – болезни, возникающие в результате нарушения числа или структуры хромосом.
·Хромосомные болезни, связанные с изменением количества хромосом (количественные/геномные хромосомные болезни как половые, так и соматические)
·Хромосомные болезни, связанные с изменением структуры одной или более хромосом (качественные хромосомные болезни)
Этиологические факторы:
1.Все виды хромосомных мутаций (делеции, дупликации, инверсии, транслокации).
2.Некоторые геномные мутации (у человека только трисомии по аутосомам, полисомии по половым хромосомам, моносомия по Х).
Классификация хромосомной патологии включает:
1.Характеристику хромосомной или геномной мутации (триплоидия, простая трисомия по хромосоме 21, частичная моносомия и т.д.);
2.Определение типа клеток, в которых возникла мутация (в гаметах – полная форма болезни, или зиготе – мозаичная форма); Мутации в соматических клетках на ранних этапах эмбриогенеза приводят к развитию мозаицизма – часть клеток организма имеет нормальный кариотип, другая
– аномальный.
3.Выявление поколения, в котором возникла мутация (спорадические, наследуемые или семейные формы).
Патогенез хромосомных болезней определяется несбалансированностью генотипа в результате геномных и хромосомных мутаций, что проявляется внутриутробной гибелью эмбрионов и плода, развитием специфических синдромов, выражающихся в нарушениях физического и психического здоровья.
Количественные хромосомные болезни с числовыми аномалиями аутосом (синдром Патау, синдром Дауна, синдром Эдвардса)
Синдром Дауна - трисомия по 21 хромосоме.(47,XX+21/47,XX+21)
По этиопатогенезу:
1.Спонтанный. Не связан с наследственными аномалиями, как правило, связан с возрастом матери (чем старше, тем выше вероятность). Нарушение мейоза – нерасхождение 21 пары.
2.Наследственный/транслокационный. Часть 21 хромосомы транслоцируется на другую (например, 15), что опять приводит к нарушению расхождения 21 (одна мутация облегчает другую).
Характерные клинические проявления синдрома Дауна
1.Внешне:
• Округлое лицо и плоская переносица.
• Монголоидный разрез глаз.
• Эпикант (кожная складка вокруг внутреннего угла глаза).
• Уплощённый затылок и малый родничок.
• Открытый маленький рот и высунутый язык.
• Маленькие уши.
• Короткая шея.
• Единственная ладонная складка, изогнутый кнутри мизинец и сандалевидная стопа (широкий промежуток между первым и вторым пальцами).
• Маленький рост.
2.Определяющие низкую жизнестойкость
•Врождённые пороки сердца (ДМЖП, ОЛП).
•Повышенный риск лейкемии (чаще ХЛЛ) и одиночных опухолей.
•Аномалия формирования иммунной системы – наследственный иммунодефицит, часто комбинированный
3.
•Задержка моторного развития.
•Умеренная или тяжелая умственная отсталость.
•Нарушение слуха, обусловленное катаральным средним отитом.
•Нарушение зрения на фоне катаракты, косоглазия, миопии.
•Болезнь Альцгеймера.
•Эпилепсия.
Синдром Патау – трисомия по 13 хромосоме (47,XX,+13 / 47,XY,+13)
Клинические проявления:
•Микроцефалия
•Полидактилия
•Расщелина губы и неба
•Низко посаженные ушные раковины
•Микроофатальмия
•Врожденные пороки сердца (ДМЖП)
•Аномалии почек
•Пороки развития органов ЖКТ
•Гипоплазия наружных половых органов (например, удвоение матки)
Синдром Эдвардса – трисомия по 18 хромосоме (47,XX+18/47,XY+18)
Клинические проявления аналогичны синдрому Патау.
Заболевания со структурными перестройками аутосом Синдром «кошачьего крика» - частичная или полная делеция короткого плеча 5 хромосомы
46,XX(5р-)/46,XY(5р-). Клиника:
•Изменение или недоразвитие гортани – характерный плач ребенка, напоминающий кошачье мяуканье, исчезает к концу 1 года жизни.
•Микроцефалия
•Умственная отсталость в стадии имбицильности
•Низкая масса и мышечная гипотония
•Лунообразное лицо с широко расставленными глазами, косоглазие
•Низко посаженные ушные раковины
Количественные хромосомные болезни с числовыми аномалиями половых хромосом Синдром Шерешевского-Тернера – полная или частичная моносомия Х-хромосомы (45, Х0)
Три группы клинических проявлений: (только женщины соответственно)
1.Гипогонадизм ( половой инфантилизм) – в пубертатном периоде аменорея, бесплодие.
2.Врожденные соматические пороки развития
- аномалии опорно-двигательного аппарата