

Виллебранда, гиперфибриногенемия. Повышается уровень ЦИК, С-РБ, возникает гиперглобулинемия, криоглобулинемия (при криоглобулинемической форме заболевания).
Диагноз. В большинстве случаев диагноз не вызывает сомнений и основывается на типичной клинической картине заболевания.
Лечение. Обязательны госпитализация и постельный режим до 3 недель. Следует избегать переохлаждения, дополнительной сенсибилизации больного лекарственными веществами и пищевыми продуктами. Из рациона исключают кофе, шоколад, цитрусовые, свежие ягоды, морепродукты. Следует избегать назначение антибиотиков, сульфаниламидов, витаминов. Антигистаминные препараты не приводят к улучшению самосувствия больных. Назначают короткие курсы глюкокортикостероидов в средних дозах, гепаринотерапию (предпочтительны низкомолекулярные гепарины – фраксипарин, клексан), плазмаферез.
Профилактика. Первичная профилактика не разработана. Профилактика рецидивов заключается в исключении провоцирующих влияний (физическое перенапряжение, охлаждение и т.д.), санации очаго инфекции.
Болезнь Верльгофа (Идиопатическая тромбоцитопеническая пурпура, аутоиммунная тромбоцитопения)
Определение. Это заболевание, при котором геморрагический синдром обусловлен тромбоцитопенией вследствие выработки аутоантител.
Эпидемиология. Частота идиопатической тромбоцитопенической пурпуры (ИТП) составляет 5,8-6,6 случаев на 100 000 населения. Женщины страдают этим заболеванием в 3 раза чаще, чем мужчины.
Этиология. Окончательно не выяснена.
Патофизиология. В основе патологического процесса лежит срыв иммунологической толерантности к собственному антигену. Под влиянием какого-либо фактора (лекарства, вируса, бактерии) изменяется антигенная струрктура тромбоцитов, они начинают восприниматься Т-лимфоцитами как чужие, активируют В-лимфоциты, которые вырабатывают аутоантитела. Иногда имеет значение генетический дефект системы Т- супрессоров, у этих лиц значительно чаще развивается любая аутоиммунная патология, в том числе аутоиммунная тромбоцитопения.
Клиника. Чаще заболевание начинается остро, а в дальнейшем характеризуется рецидивирующим либо затяжным течением. Обычно проследить какую-либо связь с перенесенной инфекцией либо другими факторами не удается, за исключением ятрогенных форм болезни, спровоцированных приемом нестероидных противовоспалительных препаратов, антибиотиков, некоторых гипотензивных препаратов и т.д.
Геморрагический синдром характеризуется кожными кровоизлияниями и кровотечениями из слизистых оболочек – петехиально-пятнистый тип кровоточивости. Кожные кровоизлияния могут иметь вид экхимозов, которые локализуются на конечностях, туловище. Часто бывают массивные синяки в местах инъекций. Петехии чаще возникают на ногах, в местах кожных складок. Появление кровоизлияний на лице, конъюнктиве, губах является неблагоприятным прогностическим признаком в отношении риска возникновения кровоизлияния в мозг. Возможны кровотечения из ЖКТ, гематурия, кровохарканье.
137
Кровотечения после хирургических манипуляций, экстракции зуба начинаются сразу после вмешательства, продолжаются несколько часов или дней, после остановки, как правило, не возобновляются, чем отличаются от отсроченных рецидивирующих кровотечений при гемофилии.
Пробы на ломкость капилляров (щипка, жгута и т.д.) часто положительны.
У 20% больных наблюдается умеренное увеличение размеров селезенки. Увеличение размеров печени не характерно.
Дополнительные методы исследования.
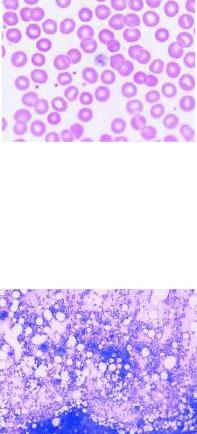
I. Общий клинический анализ крови: регистрируется тромбоцитопения (иногда вплоть до полного их исчезновения). Чаще всего геморрагический синдром развивается при уровне тромбоцитов ниже 50 Г/л. Возможны морфологические изменения тромбоцитов: увеличение их размеров, появление малозернистых «голубых» клеток, пойкилоцитоз пластинок, уменьшение количества отросчатых форм (рис. 8.3). Содержание эритроцитов и гемоглобина зависит от выраженности геморрагического синдрома. Содержание лейкоцитов обычно нормальное или незначительно увеличено. У части больных отмечается эозинофилия.
Рис. 8.3. Гигантские тромбоциты в мазке крови больнй идиопатической тромбоцитопенической пурпурой.
II. Миелограмма: чаще отмечается увеличение количества мегакариоцитов (рис. 8.4), но иногда при аутоиммунной агрессии на уровне костного мозга возможно угнетение мегакариоцитарного ростка. Часто обнаруживаются большие по размерам мегакариоциты, преобладают молодые формы, однако отшнуровка тромбоцитов может быть нарушена.
Рис. 8.4. Увеличение количества мегакариоцитов в костном мозге больной идиопатической тромбоцитопенической пурпурой.
III. Коагулограмма: нормальное или повышенное содержание плазменных факторов свертывания. Время кровотечения по Дьюке удлинено. Ретракция кровяного сгустка уменьшена. Свертываемость крови нормальная.
IV. Иммуноферментный (иммунофлуоресцентный) метод: на томбоцитах обнаруживаются антитромбоцитарные антитела (IgG).
138
Диагноз. Диагноз устанавливают на основании выявления тромбоцитопении в общем анализе крови, отсутствия причины для симптоматической тромбоцитопении, наличия на тромбоцитах антитромбоцитарных антител.
Дифференциальный диагноз. Необходимо дифференцировать ИТП с симптоматическими тромбоцитопениями. Для исключения апластической анемии и гемобластозов используют стернальную пункцию и трепанобиопсию. Системная красная волчанка в большинстве случаев сопровождается гемолитической анемией и увеличением СОЭ.
Лечение. Строго исключают из употребления все пищевые продукты и медикаменты, нарушающие агрегационные свойства тромбоцитов и вызывающие аллергизацию организма.
1.Глюкокортикостероиды. Патогенетическая терапия включает в себя назначение глюкокортикостероидов, спленэктомию, иммунодепрессанты. Лечение начинают с назначения преднизолона в дозе 1 мг/кг, эффект обычно наступает через 2-5 дней (вначале прекращается геморрагический синдром, а затем начинается рост тромбоцитов). Преднизолон в такой дозе принимают до полного эффекта, а затем постепенно медленно снижают дозу. Если через 3 месяца от начала глюкокортикостероидной терапии эффект неполный или нестабильный, возникают показания к спленэктомии.
2.Спленэктомия. По данным литературы, у 75% больных спленэктомия приводит к полному выздоровлению. Спленэктомию производят на фоне глюкокортикостероидной терапии, за 5 дней до операции дозу повышают в 2 раза, а в день операции преднизолон вводят внутривенно в дозе, увеличенной в 3 раза по сравнению с исходной пероральной. С 3- го дня после операции дозу снижают с постепенной отменой препарата.
3.Цитостатики. Неэффективность гормональной терапии и спленэктомии является показанием к назначению цитостатиков (циклофосфан) или иммунодепрессантов (азатиоприн).
4.Симптоматическая терапия. Симптоматическое лечение геморрагического синдрома включает назначение дицинона, аминокапроновой кислоты.
Трансфузии тромбоконцентрата категорически противопоказаны!
Гемофилия
Определение. Гемофилии – геморрагические диатезы из группы наследственных коагулопатий, обусловленные дефицитом определенных факторов свертывания крови.
Так, при гемофилии А отмечается дефицит VIII, при гемофилии В (болезни Кристмаса) – IX, а при гемофилии С – XI фактора.
Эпидемиология. Частота гемофилий в разных странах колеблется от 7 до 18 случаев на 100 000 населения, причем 87-94% приходится на гемофилию А.
Этиология. Наследственный дефицит или наследственная молекулярная аномалия факторов свертывания. Гемофилии А и В передаются по рецессивному сцепленному с Х-хромосомой типу наследования. Возможны ненаследственные (спорадические) случаи заболевания, обусловленные мутацией генов. Гемофилия С наследуется аутосомно-доминантно с непоной экспрессией патологического гена, поэтому одинаково часто болеют лица обоего пола.
Патогенез. Все дочери больного гемофилией А и В являются достоверными гетерозиготными носителями и передатчицами болезни. Сыновья больного гемофилией, которые получили только одну здоровую Х-хромосомы от здоровой матери здоровы и не могут передать болезнь потомству. Сыновья носительниц гемофилии имеют равные шансы
139
родиться больными или здоровыми, а дочери имеют равные шансы быть или не быть носительницами. У гетерозиготных носительниц гена гемофилии уровень факторов свертывания ниже, чем в здоровой популяции.
Классификация. Отмечается четкая зависимость между тяжестью геморрагического синдрома и уровнем дефицитного фактора свертывания.
1.Легкая форма – уровень фактора выше 5% от средней нормы.
2.Средней тяжести – от 2 до 5%.
3.Тяжелая - от 1 до 2%.
4.Крайне тяжелая – от 0 до 1%.
Клиническая картина. По симптоматике гемофилии А и В идентичны. Гемофилия С протекает обычно легче, геморрагический синдром менее выражен.
Характерен гематомный тип кровоточивости – преобладают кровоизлияния в крупные суставы (гемартрозы), глубокие подкожные, межмышечные и внутримышечные гематомы, обильные и длительные кровотечения при травмах.
Заболевание обычно дебютирует в раннем детском возрасте. Чем легче гемофилия, тем позднее проявляется геморрагический синдром.
Выделяют три разновидности поражения суставов при гемофилии:
1.Острые гемартрозы – первичные и рецидивирующие.
2.Хронические геморрагически-деструктивные остеоартрозы.
3.Вторичный ревматоидный синдром.
Острый гемартроз характеризуется внезапным (часто после незначительной травмы или спонтанно) резкой боли в суставе. Сустав увеличен в объеме, кожа над ним гиперемирована и горячая на ощупь, может определяться флюктуация. Характерно быстрое (в течение нескольких часов) ослабление боли после первой же трансфузии криопреципитата или антигемофильной плазмы и почти немедленное – при одновременной эвакуации крови из сустава с последующим введением в его полость глюкокортикостероида.
Подкожные межмышечные, субфасциальные и забрюшинные гаматомы могут достигать огромных размеров, содержать от 500 мл до 3 л крови, распространяться по продолжению, формируя глубокие затеки, прорываться в брюшную полость. Они очень болезненны, напряжены, иногда флюктуируют, вызывают анемию, лейкоцитоз, повышение температуры тела, могут нагнаиваться, вызывать некроз окружающих тканей, в том числе деструкцию костей.
Упорные почечные кровотечения наблюдаются у 14-30% больных. Возникают спонтано либо на фоне травм поясничной области, пиелонефрита.
Профузные желудочно-кишечные кровотечения могут быть спонтанными либо обусловленными приемом препаратов, вызывающих эрозирование слизистой оболочки.
Кровотечения при травмах и операциях носят отсроченный характер (возникают не сразу, а через 1-5 ч). Любые хирургические вмешательства у больных гемофилией должны выполняться только под прикрытием заместительной терапии.
Дополнительные методы исследования.
I. Выявление коагулопатии: гемофилия должна быть заподозрена во всех случаях, когда имеется гематомный тип кровоточивости. Коагулограммы: удлинение парциального тромбопластинового времени, времени свертывания крови; время кровотечения, тромбиновое и протромбиновое время нормальные.
II. Определение типа гемофилии: основой диагностики гемофилии является количественное определение уровней VIII, IX и XI факторов свертывания в крови. Перед
140
этим тщательно собирают семейный анамнез и определяют фактор, соответствующий предполагаемому типу гемофилии.
С исторической точки зрения представляют интерес коррекционные пробы, применявшиеся ранее для диагностики гемофилии:
1.Тест генерации тромбопластина по Биггс-Дугласу-Макфарлану.
2.Тест образования тромбина.
3.Коррекционная проба на базе аутокоагуляционного теста на 4-й минуте
инкубации.
Во всех этих методиках используется принцип разведения и коррекции нарушенного свертывания компонентами нормальной крови – адсорбированной сульфатом бария плазмой, выдержанной сывороткой либо адсорбированной сульфатом бария плазмой+сывороткой. Если дефект свертывания исправляется только адсорбированной сульфатом бария плазмой, в которой есть VIII фактор, но нет IX, то имеет место гемофилия А. Если дефект исправляется только нормальной сывороткой, в которой есть фактор IX, но нет VIII, то ставят диагноз гемофилии В. Если же нарушение исправляется полностью как бариевой плазмой, так и сывороткой, то вероятнее всего имеет место дефицит XI фактора - гемофилия С.
Определение типа гемофилии может быть выполнено с тестами смешивания: к плазме исследуемого больного последовательно в разных пробирках добавляют образцы плазм больных с заведомо известной формой гемофилии с почти нулевым содержанием фактора. Если смешиваются плазмы с недостатком одного и того же фактора, то коррекции гемостаза не наступает, в других случаях происходит полная нормализация свертывания.
Диагноз. Подтверждением диагноза гемофилия служит низкий уровень VIII, IX или XI фактора в сыворотке крови.
Дифференциальный диагноз. Дифференцировать приходится разные варианты гемофилии между собой, а также с болезнью Виллебранда. Разные варианты гемофилии различаются по дефицитному фактору. При болезни Виллебранда отмечается удлинение времени кровотечения и нарушение функции тромбоцитов.
Лечение. Основным методом лечения является заместительная терапия. При гемофилии А для этой цели используют очищенный концентрат фактора VIII, препараты рекомбинантного фактора VIII (до последнего времени самыми распространенными препаратами для лечения гемофилии А были кроипреципитат и свежезамороженная плазма); при гемофилии В - очищенный концентрат фактора IX, препараты рекомбинантного фактора IX, концентрат фактора протромбинового комплекса (PPSB), свежезамороженная плазма; при гемофилии С - очищенный концентрат фактора XI, препараты рекомбинантного фактора XI, свежезамороженная плазма.
При легких формах гемофилии А возможно использование десмопрессина (адиуретина), который приводит к высвобождению эндогенного фактора VIII и фактора Виллебранда и повышению в 2-3 раза уровня VIII фактора в сыворотке крови у большинства больных.
При острых гемартрозах – заместительная терапия, пункция сустава, удаление излившейся крови, введение в полость сустава глюкокортикостероидов. При повторных гемартрозах в один и тот же сустав выполняется субтотальная синовэктомия.
Осложнения после заместительной терапии. Самыми опасными осложнениями являются заражение трансмиссивными агентами и развитие ингибиторов (антител) к дефицитным факторам.
141