

796
.pdfГЛАВА 2. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ ОРГАНИЧЕСКОЙ ХИМИИ
Наиболее важным в химии является учение о превращениях веществ, в том числе об энергетике и кинетике химических реакций. Усвоение этого учения позволяет предсказать возможность и направление химических реакций, рассчитать энергетические эффекты и энергозатраты, скорость получения и выход продуктов, воздействовать на скорость химических процессов, предупреждать нежелательные реакции в тех или иных превращениях.
2.1. Энергетика химических процессов
Энергетику химических процессов изучает химическая термодинамика, с помощью которой можно определить энергетические эффекты, сопровождающие химические процессы, направление и пределы их самопроизвольного протекания.
Химическая термодинамика базируется на законах (началах) общей термодинамики.
Первый закон термодинамики является формой выражения закона сохранения энергии. Согласно этому закону энергия не может ни создаваться, ни исчезать, но может переходить из одной формы в другую. Его справедливость доказана многовековым опытом человечества. Математическим выражением I закона термодинамики является уравнение:
Q = U + A
Теплота Q, подведенная к системе, расходуется на приращение внутренней энергии системы U и на работу системы А над окружающей средой.
Внутренняя энергия системы U включает все виды энергии системы (энергию движения и взаимодействия молекул, атомов, ядер и других частиц, внутриядерную и другие виды энергии, кроме кинетической энергии системы как целого и потенциальной энергии ее положения). Она представляет собой способность системы к совершению работы или передаче теплоты. Полный запас внутренней энергии системы измерить невозможно, однако, можно определить ее изменение U при переходе системы из одного состояния в другое:
U = U1 − U2 ,
где U2 и U1 − внутренняя энергия системы в конечном и начальном состояниях. Большинство химических процессов являются изобарическими, то есть протекают при постоянном давлении.
Рассчитаем тепловой эффект Qp изобарического процесса. Из перво-
го закона термодинамики: |
|
|
|
Q = U − A |
( ) |
||
|
P |
|
1 |
|
|
|
|
|
|
61 |
|

Если на систему не действуют другие силы, кроме постоянного давления, то при протекании химического процесса единственным видом работы яв-
ляется работа расширения:
A = p V
В этом случае уравнение (1) примет вид
|
|
Q P= U |
+ |
p V |
|
|
|
|
|
|
|
Подставив в него значение U = U2 −U1 и V = V2 −V1, получим |
|
||||
Q |
P |
= U1 − U2 + pV2 − pV1 = (U2 + pV2) − (U1 + pV1) |
(2) |
||
Функция |
|
|
|
|
|
|
|
|
|
|
|
называется энтальпией си- U + pV = H |
(3) |
||||
стемы. Это одна из термодинамических функций, характеризующих систему, находящуюся при постоянном давлении. Подставив (3) в (2), полу-
чим |
Q P= H2 |
− H1 = H |
(4) |
|
В случае изобарического процесса (p = const) теплота, подведенная к системе, равна изменению энтальпии системы. Изменение энтальпии H относят к 1 молю вещества и выражают в кДж/моль.
Тепловым эффектом реакции является изменение энергии системы при протекании в ней химического процесса.
Если в результате реакции выделяется теплота, то есть понижается энтальпия системы ( H 0), реакция является экзотермической. Реакция, протекающая с поглощением энергии, то есть с повышением энтальпии
( H > 0), является эндотермической.
Тепловой эффект химической реакции рассчитывают по закону российского ученого Г.И.Гесса.
Тепловой эффект реакции зависит от природы и состояния исходных веществ и продуктов реакции, но не зависит от пути реакции, то есть от числа и характера промежуточных стадий.
Согласно следствию из закона Гесса энтальпия химической реакции равна сумме энтальпий образования продуктов реакции за вычетом суммы энтальпий образования исходных веществ с учетом стехиометрических коэффициентов:
H f0 |
х.р.= Σn i H f0 |
прод − Σn i H 0f реаг , |
где ni − стехиометрические коэффициенты в уравнении реакции, Hf −стандартная энтальпия образования 1 моля сложного вещества из простых веществ, устойчивых при 298 К и p = 101, 3 кПа. Значения Hf нескольких тысяч веществ приведены в справочниках.
В качестве примера приведем расчет теплового эффекта реакции фо-
тосинтеза: h
6H2OхлорофиллC6H12O6 + 6O2
62
Hf х.р.= Hf ( C6H12O6) + 6 Hf (O2) − 6 Hf (CO2, г) − 6 Hf (H2O, ж) = −2820.1 + 6O − 6(−393.5) − 6(214,8) = 991.7 кДж
H > 0. Реакция эндотермическая, протекает с поглощением солнечной энергии.
Второй закон термодинамики определяет направление и предел самопроизвольного протекания химических реакций, то есть выход продуктов. Чтобы понимать химические процессы и управлять ими, необходимо знать движущие силы химических реакций. Одной из движущих сил хими-
ческой реакции является энтальпия системы. Большинство экзотермических реакций протекают самопроизвольно ( H < 0). Однако известны и самопроизвольные эндотермические реакции ( H > 0), например, растворение солей. Второй движущей силой химической реакции является стремление частиц (молекул, ионов, атомов) к хаотическому движению, а системы – к переходу из менее упорядоченного состояния в более упорядоченное.
Мерой неупорядоченности состояния системы служит термодинамическая функция − энтропия S.
Изменение энтропии системы в результате протекания химической реакции вычисляют по уравнению, аналогичному уравнению Гесса:
S х.р. = Σni S 0прод − Σn S 0исх ,
где S − стандартная энтропия при 298 К, измеряемая в Дж/моль·К, приводится в справочниках. В отличие от других термодинамических функций можно определить абсолютное значение энтропии, так как энтропия при абсолютном нуле равна нулю.
В изолированных системах самопроизвольно протекают процессы с возрастанием энтропии ( S > 0). В качестве примера рассчитаем изменение энтропии в реакции фотосинтеза:
S х.р. = S ( C6H12O6) + 6S (O2) − 6S (CO2) − 6S (H2O) = 269.45 + 6∙205,03 − 6∙213.64 − 6∙69.96 = − 201.97 Дж/К = −0.202 кДж/К ,
S < 0. Энтропия системы уменьшается. В изолированных системах фотосинтез самопроизвольно протекать не может. Но если система обменивается энергией с окружающей средой, то есть является неизолированной, то возможно и обратное.
Критерием самопроизвольного протекания процесса является энергия Гиббса.
Энтальпия химического процесса состоит из двух составляющих:
H = G + Q ,
где G − свободная энергия или энергия Гиббса, являющаяся частью внутренней энергии системы, за счет которой совершается работа; Q − связанная (обесцененная) энергия, которая не превращается в полезную рабо-
63
ту, а рассеивается в окружающую среду в виде теплоты Q = T S; отсюдаН = G + T S, то есть
G = H − T S
Это уравнение является основным в химической термодинамике. Н− энтальпийный фактор, T S − энтропийный фактор.
Энергию Гиббса химического процесса можно вычислить несколькими способами:
– по основному уравнению химической термодинамики; по уравнению, аналогичному закону Гесса
G 0х.р. = Σni |
G0 |
прод − Σni G0реаг ; |
–по величине ЭДС гальванического элемента, в котором эта реакция используется для получения электрического тока G = − nFE, где n − заряд ионов, F − число Фарадея, Е − ЭДС;
–по величине константы равновесия G = − RT lnK.
Химическая реакция принципиально возможна, если энергия Гиббса уменьшается ( G < 0). Если энергия Гиббса возрастает ( G > 0), реакция самопроизвольно протекать не может. Наконец, если G = 0, то система находится в равновесии, реакция обратима. В качестве примера рассчитаем изменение энергии Гиббса реакции фотосинтеза при 298 К:
G = Н − T S = 991.7 − 298 · (− 0.202) = 1051.9 кДж, G >0. В изо-
лированной системе самопроизвольное протекание реакции фотосинтеза невозможно.
Применение законов термодинамики в биологии
Классическая химическая термодинамика имеет дело с замкнутыми системами, находящимися в равновесии. Живые организмы − открытые системы. Клетка в равновесном состоянии − мертвая клетка. Реакции, протекающие в живой клетке, как и любые другие реакции, можно разделить на две категории.
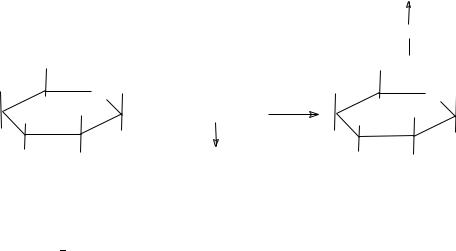
Для реакций, сопровождающихся усложнением молекул (фотосинтез, синтез липидов, белков, углеводов, нуклеиновых кислот и др.) G > 0. Самопроизвольно эти процессы идти не могут. Для их осуществления требуется внешний источник свободной энергии (световая энергия, АТФ и другие). Таким образом, осуществляется термодинамический контроль подобных реакций.
Для реакций, сопровождающихся расщеплением молекул, G < 0. Они энергетически выгодны, однако, скорости их часто пренебрежимо малы. Например, реакция фосфорилирования глюкозы:
64
|
|
|
|
|
|
|
|
|
O |
|
|||
CH2OH |
|
|
|
|
|
|
HO |
|
P |
|
OH |
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
CH O |
|
||||||
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
H |
O H |
|
|
OH |
H |
|
|
O H |
|
||||
H |
|
|
|
|
|
||||||||
H |
+ HO |
|
|
|
|
|
H |
|
H |
|
|||
|
|
|
|
|
|
|
|||||||
OH |
|
|
P OH |
OH |
|
|
|
|
+ H2O |
||||
OH |
OH |
|
|
O |
OH |
|
|
|
OH |
|
|||
H |
OH |
|
|
H |
|
OH |
|
||||||
|
|
|
|
|
|
|
|
||||||
Для протекания подобных реакций необходимо присутствие органических катализаторов − ферментов. Такие реакции подчиняются кине-
тическому контролю.
2.2. Кинетика химических процессов
Как показано ранее, химическая термодинамика позволяет предсказать принципиальную возможность или невозможность самопроизвольного течения химических реакций. Однако знания рассмотренных закономерностей недостаточно, чтобы предсказать реальную возможность химической реакции, определить ее скорость и механизм, возможность управлять процессом. Скорость реакции часто не связана со значением ее энергии Гиббса. Например, термодинамическая вероятность реакции окисления водорода до воды:
H2 + ½ O2 = H2O ж |
G2980 = −237. 2 кДж/моль |
|
значительно выше вероятности реакции нейтрализации с образова- |
||
нием воды: |
|
|
H+ + OH−= Η2Οж |
G2980 |
= −79..9 кДж/моль |
Однако первая реакция без катализатора практически не идет, а вторая протекает мгновенно.
В органической химии кинетику изучают с целью доказательства механизма реакции, устанавливая лимитирующие стадии химических процессов. Скорости химических реакций, факторы, влияющие на скорость и механизм химических реакций, изучает химическая кинетика.
Скорость химической реакции определяется изменением концентрации реагентов в единицу времени.
Различают среднюю и мгновенную скорости химического процесса.
Средняя скорость определяется как |
|
< v > = (c2 − c1) / (t2− t1) = − c/ t |
моль/ л . с , |
где с2 и с1 − концентрации исходного вещества в моменты времени t2
65

и t1, знак «минус» означает, что концентрация исходного вещества уменьшается в ходе реакции.
Мгновенная (истинная) скорость − это скорость реакции в данный
момент: |
v = −dc/ dt |
|
Скорость химической реакции связана с концентрациями реагирующих веществ кинетическим уравнением − законом действующих масс для химической кинетики:
aA + bB  cC + dD
cC + dD
v = kCAa C bB ,
где CA и CB − концентрации реагентов, a и b − стехиометрические коэффициенты в уравнении реакции, k − константа скорости реакции, то есть скорость реакции при CA = CB = 1 моль/л. Константа скорости реакции не зависит от концентрации реагентов, а зависит от их природы и температуры.
Порядок химической реакции равен сумме порядков по реагентам в кинетическом уравнении n = Σ ni. Например,
H2 + Cl2  2HCl (газ), v = CH2 · CI2 , n = 1 + 1 = 2.
2HCl (газ), v = CH2 · CI2 , n = 1 + 1 = 2.
Впростых одностадийных реакциях порядок реакции определяется стехиометрическими коэффициентами в уравнении реакции. Для многостадийных реакций порядки реакции по реагентам, как правило, не совпадают со стехиометрическими коэффициентами и определяются экспериментально по скорости реакции самой медленной (лимитирующей) стадии.
Взависимости от числа реагирующих частиц, участвующих в элементарном акте, реакции различаются по молекулярности.
Мономолекулярными реакциями являются всевозможные процессы
разложения A B + C + , например,
CH3CH2OH H2C CH2 + H2O
H2C CH2 + H2O
Кбимолекулярным реакциям относят реакции типа A + B C + D +
или 2A B + C + , например,
CH3COOC2H5 + H2O CH3COOH + C2H5OH
CH3COOH + C2H5OH
Если в элементарном акте принимают участие три молекулы, реакция является трехмолекулярной. Реакции с молекулярностью выше трех неизвестны.
Рассмотрим далее влияние температуры на скорость реакции. Повышение температуры ускоряет большинство химических реакций. Со-
66

гласно правилу Вант-Гоффа повышение температуры на 10 К повышает скорость реакции в 2 4 раза:
v2/ v1 = (T2 − T1) / 10 ,
где v2 и v1 − скорости реакции при температурах T2 и T1 , − температурный коэффициент реакции, = 2 4. Точность правила Вант-Гоффа невысока, им можно пользоваться лишь для ориентировочных расчетов. Более точная зависимость скорости реакции от температуры описывается
уравнением Аррениуса:
RT ,
где k− константа скорости реакции, k0 − предэкспоненциальный множитель, e − основание натурального логарифма, Ea − энергия активации, R− универсальная газовая постоянная, T − температура, К.
Энергия активации. Чтобы дать ответ на вопрос, что такое энергия активации и как она влияет на скорость реакции, рассмотрим в общем виде механизм реакции.
В ходе реакции разрушаются одни и возникают другие молекулы, происходит изменение химических связей, перераспределение электронной плотности. Полное разрушение всех старых химических связей требует больших затрат энергии. Как показали исследования, в ходе реакции система проходит через переходное состояние − образование активированно-
го комплекса.
Ход реакции AB + CD AС + BD можно представить схемой:
A |
|
|
B |
A......B |
A |
B |
|||
|
|||||||||
|
|
|
|
. . |
|
|
|
|
|
|
+ |
|
|
. . |
|
|
+ |
|
|
|
|
|
. . |
|
|
|
|
||
|
|
|
|
|
|
||||
C |
|
|
D |
C......D |
C |
D |
|||
|
|
||||||||
исходные |
активированный |
продукты |
|||||||
молекулы |
комплекс |
|
реакции |
||||||
В активированном комплексе старые связи еще не разорваны, но уже ослаблены, новые связи наметились, но еще не образовались. Время существования его невелико ( 10−13 с). При распаде комплекса образуются либо продукты реакции, либо исходные вещества. Энергия, необходимая для пе-
рехода вещества в состояние активированного комплекса, называется «энергией активации».
67
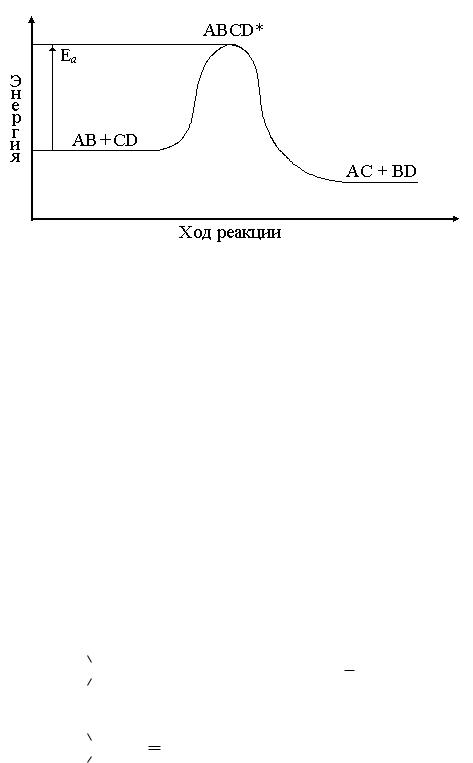
Рис.2.1.
Энергетическая диаграмма хода реакции с образованием активированного комплекса.
Частицы называются активными, если их энергия равна или выше энергии активированного комплекса. С ростом температуры растет доля активных частиц и возрастает скорость реакции.
Предэкспоненциальный множитель. Из уравнения Аррениуса следу-
ет, что k = k0 при Ea = 0, то есть можно предположить, что при Ea = 0 каждое столкновение реагирующих частиц приводит к химической реакции. Но, как показывает опыт, для большинства молекул k0 < z (где z − число столкновений частиц в единицу времени), то есть не каждое столкновение даже активных частиц приводит к реакции. Необходимо еще одно условие протекания реакции − ориентация молекул реагентов, благоприятствующая перераспределению электронной плотности. Например, при взаимодействии H2O и CO возможна их различная ориентация друг относительно друга:
H
O + C O
O  O
O C
C O + H H
O + H H
H
H
O + O C  H2O + CO
H2O + CO
H
Осуществлению реакции благоприятствует первый вариант ориентации молекул.
Таким образом, предэкспоненциальный множитель отражает частоту столкновений и ориентацию реагирующих частиц. Реакция возможна при достаточной энергии и надлежащей ориентации частиц.
2.3. Механизмы органических реакций
68

В уравнениях химических реакций, которыми мы пользуемся для выражения химических процессов, указываются только лишь исходные вещества и продукты реакции:
A + B C + D
C + D
и не показано, каким образом исходные вещества превращаются в продукты реакции, то есть они не раскрывают механизма реакции. Под
механизмом реакции понимаются промежуточные стадии, через которые происходит превращение исходных веществ в продукты реакции.
Естественно, в химических реакциях всегда происходит разрыв химической связи в исходных веществах и образование новой химической связи в продуктах реакции. Промежуточные стадии реакции зависят от характера разрыва химической связи в исходных веществах. Различают два типа разрыва ковалентной связи:
а) гомолитический разрыв:
C : R |
h |
C |
. + R. |
|
При гомолитическом распаде ковалентной связи образуются проме-
жуточные радикалы. Радикалы − это электронейтральные частицы, имеющие орбиталь с одиночным электроном. Название радикалам дается по названию углеводорода с заменой суффикса «ан» на суффикс «ил», например, CH3 − метил, CH3 − СН2 − этил и т.д. Часто они называются тривиально: CH2 = CH − винил, C6H5 − фенил и т.д. Гомолитическому распаду связи способствуют высокая температура, УФ-облучение, наличие свободных радикалов в реакционной массе.
б) гетеролитический разрыв:
 C :
C : R
R 
 C−: + R+
C−: + R+
карбанион
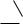 C
C  : R
: R 
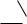 C+ + :R−
C+ + :R−
карбокатион
При гетеролитическом разрыве промежуточно образуются заряженные частицы − ионы. Гетеролитическому распаду связи способствуют кислотные или щелочные катализаторы.
Этим двум вышеуказанным типам разрыва ковалентной связи соответствуют два типа механизма органических реакций − радикальный (промежуточное образование радикалов) и ионный (промежуточное образование ионов).
В химических реакциях условно различают атакуемую молекулу − субстрат и атакующую молекулу − реагент [9]. Принято классифициро-
69

вать химические реакции по реагентам. Реагенты по характеру подразделяются на нуклеофилы (Nucleous − ядро, filio − любить, любовь; любовь к положительно заряженному ядру атома) и электрофилы (electron −электрон, filio − любить, любовь; любовь к отрицательно заряженному электрону).
Нуклеофилы − это реагенты, способные предоставлять электронную пару на образование ковалентной связи с высших занятых молекулярных орбиталей (ВЗМО). ВЗМО условно можно обозначить .. . Другими словами, они являются донорами электронной пары (ДЭП). Нуклеофилы подразделяются на:
а) n-ДЭП, являющиеся молекулами, атомами и анионами, имеющими, по крайней мере, одну свободную пару электронов на р- или sp- гибридной атомной орбитали (АО): RX, X−, R2O, RO−, R2S, RS−, R3N, R3P, R3As, R3C−, CO и другие. Одновременно они являются основаниями Льюиса;
б) -ДЭП, являющиеся молекулами или анионами, содержащими пару электронов на π-молекулярной орбитали (МО): этилен, бутадиен, ацетилен, бензол и др.;
в) -ДЭП, представляющие собой молекулы, содержащие пару электронов на σ-МО, в качестве которой могут служить только электроны свя-
зи: С−С, или С−Н; R3C−CR3; R3C−H, H−H и др.
Электрофилы− это реагенты, которые в ходе реакции приобретают электронную пару (акцепторы электронной пары − АЭП) на низшую свободную молекулярную орбиталь (НСМО), которую условно можно обозначить 
 . Они классифицируются на:
. Они классифицируются на:
а) -АЭП, являющиеся молекулами, образующими с ДЭП -МО: тетрацианэтилен, тринитробензол и др.;
б) -АЭП, которыми могут быть атомы, молекулы или катионы, образующие с ДЭП преимущественно -МО: Ni, BF3, H+, R3C+, Fe2+, Pt2+ и др. Такие частицы являются одновременно кислотами Льюиса.
Следует отметить, что аналогичную классификацию можно провести также и у субстратов (атакуемые молекулы).
Известно, что концепция окисления и восстановления является частным случаем обобщенной концепции электрофильности и нуклеофильности. Поэтому все окислители являются электрофилами и все восстановители – нуклеофилами.
Каждый механизм имеет свой символ. Он характеризует:
а) тип реакции по направлению (замещение− substitution S, присо-
единение − addition A, отщепление − elimination E);
б) характер (свойство) промежуточной частицы − Nucleofil (N), Electrofil (E) и радикал (R);
70