

количества бластных клеток) и выявляется лимфоидная их направленность, оптимальными являются комбинации препаратов, содержащих винкристин и преднизолон, такие как восьмидневный курс VRP — винкристин, доксорубицин, преднизолон, COAP — циклофосфан, онковин, адриабластин, преднизолон.
Прогноз Выживаемость при хронических миелопролиферативных болезнях при грамотной
своевременно начатой терапии составляет около 20 лет. Если лечение не проводится, то пациент умирает в течение полутора лет.
Прогноз при хроническом миелолейкозе зависит от множества факторов, определяющим из которых является момент начала лечения (в хронической фазе, фазе активации или в период бластного криза). В качестве неблагоприятных прогностических признаков хронического миелолейкоза рассматривают значительное увеличение печени и селезенки (печень выступает из-под края реберной дуги на 6 и более см, селезенка – на 15 и более см), лейкоцитоз свыше 100x109/л, тромбоцитопению менее 150x109/л, тромбоцитоз более 500х109/л, повышение уровня бластных клеток в периферической крови до 1% и более, повышение суммарного уровня промиелоцитов и бластных клеток в периферической крови до 30% и более.
Вероятность неблагоприятного исхода при хроническом миелолейкозе возрастает по мере увеличения количества признаков. Причиной гибели становятся инфекционные осложнения или тяжелые геморрагии. Средняя продолжительность жизни пациентов с хроническим миелолейкозом составляет 2,5 года, однако при своевременном начале терапии и благоприятном течении заболевания этот показатель может увеличиваться до нескольких десятков лет.
Вцелом переход заболевания в фазу бластного криза сочетается с крайне низкими показателями выживаемости, как правило, не превышающими 6 мес у больных с миелоидными и недифференцированными вариантами.
Профилактика
Всвязи с невозможностью в настоящее время выделить этиологические факторы заболевания разработка конкретных рекомендаций по первичной профилактике ХМЛ в настоящее время невозможна.
После выявления заболевания наиболее важными факторами сохранения жизни и здоровья пациентов являются как можно более быстрое начало терапии ИТК и строгая приверженность пациента к выполнению рекомендаций по лечению и мониторингу ответа на терапию.
Современное медикаментозное лечение пациентов с ХМЛ является высокоэффективным у подавляющего большинства пациентов. Имеющиеся рекомендации по контролю нежелательных явлений ИТК и возможность альтернативного выбора препаратов позволяет практически полностью сохранить физическое состояние и повседневный уровень активности.
• Рекомендуется всем пациентам с ХМЛ профилактика фоновых предопухолевых заболеваний и состояний, приверженность здоровому образу жизни, исключение хронических интоксикаций, ограничение контакта с вредными производственными факторами, участие в мероприятиях диспансеризации
• Рекомендуется всем пациентам с ХМЛ диспансерное наблюдение для оценки эффективности терапии, контроля побочных действий и лекарственной токсичности.
42. Геморрагические диатезы. Механизмы первичного и вторичного гемостаза. Классификация. Типы кровоточивости и их клинико-лабораторные критерии.
Определение - Это группа болезней и патологических состояний наследственного или приобретенного
характера, общим проявлением которых является геморрагический синдром: склонность к рецидивирующим интенсивным длительным, чаще всего множественным, кровотечениям и кровоизлияниям Механизмы первичного и вторичного гемостаза.
1)первичный гемостаз (тромбоцитарно-сосудистая реакция)- благодаря этому механизму происходит остановка кровотечения из мелких сосудов с низким артериальным давлением:
- спазм сосудов рефлекторного характера и связанный с БАВ -эндотелий изменяет свой заряд с отрицательного на положительный -активация тромбоцитов под действием коллагена и ф. Виллебранда -адгезия тромбоцитов - агрегация тромбоцитов -ретракция тромбоцитарного тромба
2)вторичный гемостаз (коагуляционный)- свертывание крови – это цепной ферментативный процесс ,в котором последовательно происходит активация факторов свертывания и образование их комплексов. Сущность свертывания крови заключается в переходе растворимого белка крови фибриногена в нерастворимый фибрин, в результате чего образуется прочный фибриновый тромб.
Классификация:
1)Геморрагические диатезы, обусловленные дефектом тромбоцитарного звена:
•недостаточность количества тромбоцитов;
•функциональная неполноценность тромбоцитов;
•сочетание количественной и качественной патологии тромбоцитов.
2) Геморрагические диатезы, обусловленные дефектом прокоагулянтов (гемофилии):
•недостаточное их количество, необходимое для формирования фибрина;
•недостаточная функциональная активность отдельных прокоагулянтов;
•наличие в крови ингибиторов отдельных прокоагулянтов.
3)Геморрагические диатезы, обусловленные дефектом сосудистой стенки:
•врожденные;
• приобретенные.
4)Геморрагические диатезы, обусловленные избыточным фибринолизом:
• эндогенным (первичным и вторичным); • экзогенным.
5)Геморрагические диатезы, обусловленные сочетанием нарушений различных компонентов системы гемостаза (болезнь Виллебранда, ДВС-синдром и др.).
В процессе свертывания крови принимают участие следующие компоненты:
1)прокоагулянты (плазменные и пластиночные факторы свертывания), взаимодействие которых приводит к образованию сгустка фибрина;
2)антикоагулянты, препятствующие свертыванию крови (антитромбин III, гепарин);
3)ингибиторы антикоагулянтов (антигепариновые факторы тромбоцитов, эритроцитов, сыворотки; гепариназа), снижающие противосвертывающее действие антикоагулянтов.
Фибринолитическая системаявляется резервной, ей принадлежит огромная роль в процессе декоагуляции, растворения сгустков фибрина и реканализации сосуда.
Типы кровоточивости и их клинико-лабораторные критерии:
1) Гематомный — характеризуется болезненными напряженными кровоизлияниями как в мягкие ткани, так и в суставы — типичен для гемофилии А и В.
2)Петехиальнопятнистый (синячковый) — характерен для тромбоцитопений, тромбоцитопатий и некоторых других нарушений свертываемости крови (исключительно редких): гипо- и дисфибриногенемий, наследственного дефицита факторов X и II, иногда
VII.
3)Васкулитно-пурпурный тип характеризуется геморрагиями в виде симметричной мелкоточечной сыпи и/или пальпируемой пурпурой, не связанной с тромбоцитопенией. Возможно поражение внутренних органов (присоединение нефрита, кишечных кровотечений и др.). Наблюдается при васкулитах.
4)Ангиоматозный тип наблюдается при телеангиэктазиях, болезни Рандю– Ослера, ангиомах, артериовенозных шунтах. Характеризуется упорными строго локализованными и привязанными к локальной сосудистой патологии геморрагиями.
5)Смешанный синячково-гематомный тип — характеризуется сочетанием петехиальнопятнистой кровоточивости с появлением отдельных больших гематом (забрюшинных, в
стенке кишечника и т.д.) при отсутствии поражения суставов и костей (отличие от гематомного типа), либо с единичными геморрагиями в суставы: синяки могут быть обширными и болезненными. Такой тип кровоточивости наблюдается при болезни Виллебранда, ДВС-синдроме, передозировке антикоагулянтов и тромболитиков, при появлении в кpови иммунных ингибиторов факторов VIII или IX.
43. Понятие о коагулопатии. Гемофилия. Этиология. Патогенез. Клиническая картина. Прогноз. Лечение.
Коагулопатия - патологическое состояние организма, обусловленное нарушениями свёртываемости крови.
Гемофилия — наследственное заболевание свертывающей системы крови, возникающее в результате дефицита фактора свертывания крови VIII (FVIII) — гемофилия А, или фактора свертывания крови IX (FIX) — гемофилия B.
Этиология и патогенез:
Гемофилия передается по X-сцепленному рецессивному пути наследования. Примерно у 70% больных имеется положительный семейный анамнез по заболеванию. Причиной гемофилии являются мутации гена, кодирующего FVIII (Xq28), или гена, кодирующего FIX (Xq27). В 30-35% случаев возможны спорадические мутации без наличия семейного анамнеза заболевания.
Подавляющее большинство больных гемофилией— мужчины.
Классификация:
Клиническая картина:
Основное проявление гемофилии – кровотечения и кровоизлияния, возникающие спонтанно или вследствие травмы. ГА и ГВ имеют схожую клиническую картину.
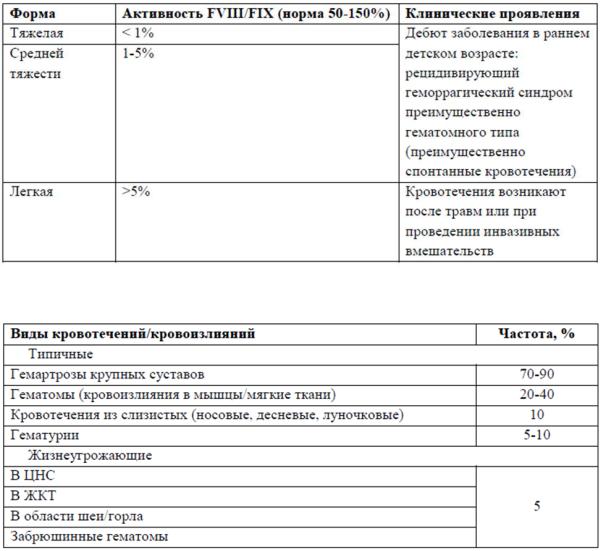
Для тяжелой формы гемофилии характерно появление геморрагического синдрома на первом году жизни с начала активного периода у ребенка (гематомы мягких тканей, посттравматические кровотечения из слизистых, гемартрозы). Поражаются в основном крупные суставы: коленные, голеностопные, локтевые и тазобедренные.

Гемофилия средней тяжести имеет сходные проявления. Первые признаки, как правило,
развиваются после года. У пациентов с активностью факторов более 2% реже возникают кровоизлияния в суставы, забрюшинные гематомы, гематурии. Наиболее типичны посттравматические гематомы и длительные кровотечения, особенно при травмах слизистых оболочек.
Легкая гемофилия может никак не проявляться на протяжении всей жизни. Геморрагический синдром обычно возникает вследствие значительных травм или при хирургическом лечении. Поражение опорно-двигательного аппарата встречается чрезвычайно редко.
Диагностика:
Для установления диагноза гемофилии у пациента с наличием геморрагического синдрома в анамнезе или при отягощенном семейном анамнезе используются критерии диагноза гемофилии (диагноз устанавливается при наличии как минимум двух из трех критериев):
●отсутствие приобретенных коагулопатий;
●снижение активности FVIII/FIX ниже 50%;
●наличие мутаций генов FVIII или FIX.
По результатам лабораторных исследований наблюдается удлинение времени свертывания крови, АЧТВ, снижение активности VIII или IX фактора.
Жалобы и анамнез
Диагностика гемофилии начинается с выявления наличия геморрагического синдрома в анамнезе у пациента и членов семьи.
Диагностика гемофилии у плода возможна на ранних сроках беременности (8– 12 нед), планирование беременности у матери-носителя гена гемофилии возможно методом ЭКО; имплантацию эмбриона выполняют только после диагностики.
Лечение:
Основное лечение — специфическая заместительная терапия концентратами факторов: адекватное замещение недостающего фактора свертывания. Существует две тактики лечения: терапия «on demand» (по требованию при кровотечении) и регулярная длительная терапия в целях профилактики кровотечений.
Необходимо использовать очищенные, вирусинактивированные препараты, изготовленные из донорской плазмы человека (концентрат FVIII, концентрат FIX, концентрат FVIII + фактор фон Виллебранда) или рекомбинантные концентраты факторов свертывания (Октоког-альфа, Мороктоког-альфа, Нонаког-альфа). В настоящее время нет оснований для предпочтения друг другу плазматических или рекомбинантных факторов свертывания. Препараты вводятся внутривенно. Применение свежезамороженной плазмы или криопреципитата разрешено только в исключительных случаях!
Для надежного гемостаза активность факторов должна быть увеличена до 15–30%, перед хирургическими вмешательствами — до 50–100%. Лечение должно быть начато как можно раньше после травмы или появления признаков кровотечения. Современное лечение гемофилии основано на принципе «домашнего лечения». Для соблюдения этого принципа должны выполняться следующие условия:
- наличие гемостатического препарата у пациента (препарат там же, где и пациент), -решение о начале заместительной терапии принимает пациент и/или его родственники в
соответствии с рекомендациями гематолога, пациент и/или его родственники должны знать правила применения и хранения препарата.
В качестве дополнительных мер гемостаза могут использоваться следующие средства:
•десмопрессин (DDAVP) с целью увеличения активности VIII фактора только у пациентов с легкой формой гемофилии А. Существуют формы в виде назального спрея;
•ингибиторы фибринолиза используются как дополнение к заместительной терапии. Противопоказаны при почечных кровотечениях;
•местные гемостатические средства также применяются как дополнение к заместительной терапии. Используются при операциях на паренхиматозных органах, экстракциях зубов.
Профилактика:
Пациенты с гемофилией должны наблюдаться группой специалистов различного профиля, включающей врача-гематолога, врача-педиатра, врача-травматолога-ортопеда, врачастоматолога, врача-физиотерапевта, врача ЛФК, медицинского психолога, имеющих опыт работы с больными гемофилией.
Осмотр пациентов врачом-гематологом, врачом-травмотологом-ортопедом и врачомстоматологом должен проводиться не менее 2-х раз в год; остальными специалистами - по необходимости. Целесообразно проведение диспансеризации пациентов 1 раз в год в специализированном центре нарушений гемостаза, если центр располагает достаточной
клинико-лабораторной |
|
|
|
|
базой. |
|
Диспансерное |
наблюдение |
за |
пациентами |
с |
гемофилией |
включает: |
Динамический мониторинг состояния пациента с оценкой наличия нежелательных явлений при проведении заместительной терапии: появление ингибитора к фактору свертывания крови, индивидуальная непереносимость препарата, вирусная контаминация, изменения психологического или социального статуса пациента, оценка состояния периферической венозной системы.
Лечение осложнений гемофилии: коррекция дефицита железа, ингибиторов.
Выявление сопутствующих заболеваний, особенно заболеваний зубов, полости рта, ЖКТ, ЛОР-органов, патологии сердечно-сосудистой системы и др. и направление к профильным специалистам.
Всех пациентов с гемофилией рекомендовано регистрировать и наблюдать в специализированном центре.
44. Тромбоцитопении. Болезнь Верльгофа. Определение. Этиология. Патогенез. Клиническая картина. Диагностика. Лечение.
Тромбоцитопенией называется состояние, при котором количество тромбоцитов в периферической крови снижается менее 150 × 109/л. Тромбоцитопения может являться как самостоятельным заболеванием, так и симптомом патологий других органов. Данный вид заболевания получил название «вторичная тромбоцитопения».
Первичная иммунная тромбоцитопения (идиопатическая тромбоцитопеническая пурпура, болезнь Верльгофа) — это аутоиммунное заболевание, обусловленное выработкой антител к структурам мембраны тромбоцитов и их предшественников — мегакариоцитов (МКЦ), что вызывает не только повышенную деструкцию тромбоцитов, но и неадекватный тромбоцитопоэз, характеризуется изолированной тромбоцитопенией ниже 100×109/л и наличием/отсутствием геморрагического синдрома различной степени выраженности.
Этиология: неизвестна. Триггерные факторы: инфекции (преимущественно вирусные), беременность, стресс, хирургические манипуляции , физическая нагрузка, прививки. Патогенез заключается в выработке аутоиммунных антител, относящися к классу IgG, к определенным гликопротеинам тромбоцитов и мегакариоцитов. Основной мишенью, против которой направлены антитромбоцитарные антитела, является гликопротеин GP IIb/IIIa, реже встречаются анти GPIb/IX антитела и еще реже — антитела, направленные против других антигенов поверхности тромбоцитов или антитела множественной специфичности. Образуется патологический комплекс антиген–антитело, фиксирующийся своим Fc-фрагментом иммуноглобулина к Fc-рецептору макрофагов и дендритических клеток ретикулоэндотелиальной системы. Разрушение комплекса происходит в основном в селезенке, реже в печени и лимфатических узлах (ЛУ), а также путем цитотоксического и комплементзависимого лизиса Классификация:
По течению:
–острые (продолжающиеся менее 3 мес.)
–персистирущие (от 3 до 12 мес)
–хронические (более 12 мес)
По периоду болезни:
– обострение (криз)