


Neoplasms, Cysts, and Tumor-Like Lesions
730
Selected References
Cranial Nerve Anatomy
Upper Cranial Nerves
Schwob JE et al: The stem and progenitor cells of the mammalian olfactory epithelium: Taking poietic license. J Comp Neurol. 525(4):1034-1054, 2017
Tsutsumi S et al: Visualization of the olfactory nerve using constructive interference in steady state magnetic resonance imaging. Surg Radiol Anat. 39(3):315-321, 2017
Elder C et al: Isolated abducens nerve palsy: update on evaluation and diagnosis. Curr Neurol Neurosci Rep. 16(8):69, 2016
Miller SR et al: Evidence for a Notch1-mediated transition during olfactory ensheathing cell development. J Anat. 229(3):369-83, 2016
Seeburg DP et al: The role of imaging for trigeminal neuralgia: a segmental approach to high-resolution MRI. Neurosurg Clin N Am. 27(3):315-26, 2016
Tantiwongkosi B et al: Imaging of ocular motor pathway. Neuroimaging Clin N Am. 25(3):425-38, 2015
Yu F et al: Advanced MR imaging of the visual pathway. Neuroimaging Clin N Am. 25(3):383-93, 2015
Lower Cranial Nerves
Haller S et al: Imaging of neurovascular compression syndromes: trigeminal neuralgia, hemifacial spasm, vestibular paroxysmia, and glossopharyngeal neuralgia. AJNR Am J Neuroradiol. 37(8):138492, 2016
Ong CK et al: The glossopharyngeal, vagus and spinal accessory nerves. Eur J Radiol. 74(2):359-67, 2010
Schwannomas
Schwannoma Overview
Ando T et al: Comparison between MR imaging findings of intracranial and extracranial schwannomas. Clin Imaging. 42:218223, 2017
Antonescu CR et al: Schwannoma. In: Louis DN et al (eds), WHO Classification of Tumours of the Central Nervous System. Lyon, France: International Agency for Research on Cancer, 2016, pp 214-218
Skolnik AD et al: Cranial nerve schwannomas: diagnostic imaging approach. Radiographics. 36(5):1463-77, 2016
Vestibular Schwannoma
Caltabiano R et al: A mosaic pattern of INI1/SMARCB1 protein expression distinguishes schwannomatosis and NF2-associated peripheral schwannomas from solitary peripheral schwannomas and NF2-associated vestibular schwannomas. Childs Nerv Syst. 33(6):933-940, 2017
Coffey N et al: Imaging findings in sensorineural hearing loss: a pictorial essay. Can Assoc Radiol J. 68(2):106-115, 2017
Håvik AL et al: Genetic landscape of sporadic vestibular schwannoma. J Neurosurg. 1-12, 2017
Schulze M et al: Improvement in imaging common temporal bone pathologies at 3 T MRI: small structures benefit from a small field of view. Clin Radiol. 72(3):267.e1-267.e12, 2017
Daultrey CR et al: Size as a risk factor for growth in conservatively managed vestibular schwannomas: the Birmingham experience. Otolaryngol Clin North Am. 49(5):1291-5, 2016
Trigeminal Schwannoma
Agrawal A et al: Extracranial trigeminal schwannomas: a retrospective analysis. J Maxillofac Oral Surg. 16(2):164-169, 2017
Agarwal A: Intracranial trigeminal schwannoma. Neuroradiol J. 28(1):36-41, 2015
Facial Nerve Schwannoma
McRackan TR et al: Primary tumors of the facial nerve. Otolaryngol Clin North Am. 48(3):491-500, 2015
Schwannomas of Other Intracranial Nerves
Zhang Q et al: Intraand extramedullary dumbbell-shaped schwannoma of the medulla oblongata: a case report and review of the literature. World Neurosurg. 98:873.e1-873.e7, 2017
Shin RK et al: Transient ocular motor nerve palsies associated with presumed cranial nerve schwannomas. J Neuroophthalmol. 35(2):139-43, 2015
Suthar PP et al: Isolated hypoglossal nerve schwannoma: an uncommon presentation of schwannoma. J Clin Diagn Res. 9(10):TJ01-2, 2015
Parenchymal Schwannomas
Luo W et al: Intracranial intraparenchymal and intraventricular schwannomas: report of 18 cases. Clin Neurol Neurosurg. 115(7):1052-7, 2013
Melanotic Schwannoma
Agarwalla PK et al: Pigmented lesions of the nervous system and the neural crest: lessons from embryology. Neurosurgery. 78(1):142-55, 2016
Spina A et al: Intracranial melanotic schwannomas. J Neurol Surg A Cent Eur Neurosurg. 76(5):399-406, 2015
Schwannomatosis
Kehrer-Sawatzki H et al: The molecular pathogenesis of schwannomatosis, a paradigm for the co-involvement of multiple tumour suppressor genes in tumorigenesis. Hum Genet. 136(2):129-148, 2017
Faucett EA et al: A diagnostic dilemma: multiple primary intracranial tumors without vestibular schwannomas. Ann Otol Rhinol Laryngol. 125(11):938-942, 2016
Neurofibromas
Perry A et al: Neurofibroma. In: Louis DN et al (eds), WHO Classification of Tumours of the Central Nervous System. Lyon, France: International Agency for Research on Cancer, 2016, pp 219-221
Röhrich M et al: Methylation-based classification of benign and malignant peripheral nerve sheath tumors. Acta Neuropathol. 131(6):877-87, 2016

Chapter 24
731
Lymphomas and Hematopoietic
and Histiocytic Tumors
The spectrum of hematopoietic neoplasms and tumorlike disorders that affect the CNS ranges from frankly malignant neoplasms like diffuse large B-cell lymphoma to nonneoplastic lesions such as extramedullary hematopoiesis.
For purposes of discussion, this chapter is divided into three major sections:
(1) lymphomas and related disorders, (2) histiocytic tumors, and (3) hematopoietic tumors and tumor-like lesions (leukemias, plasma cell neoplasms, and extramedullary hematopoiesis). We begin our discussion with the largest group, lymphomas and related disorders.
Histiocytic tumors occasionally involve the CNS. These neoplasms and nonneoplastic tumor-like masses are composed of histiocytes that are microscopically identical to their extracranial counterparts. Both Langerhans cell histiocytosis and non-Langerhans histiocytoses such as ErdheimChester disease, Rosai-Dorfman disease, juvenile xanthogranuloma, and histiocytic sarcoma are considered in the second section.
Lastly, we then turn our attention to hematopoietic tumors and tumor-like lesions. Although the most recent WHO classification includes only malignant lymphomas and histiocytic tumors in the group of hematopoietic neoplasms, we include leukemia in this chapter even though CNS involvement is almost always secondary to systemic disease.
Plasma cell myeloma and solitary plasmacytoma affecting the skull and brain are also usually secondary to extracranial disease, but they are a form of mature B-cell neoplasms and hence are included here rather than in Chapter 27 on metastases. We conclude the chapter with a brief discussion of extramedullary hematopoiesis—benign, nonneoplastic proliferations of blood-forming elements—which can appear virtually identical to malignant hematopoietic neoplasms.
Lymphomas and Related Disorders 731
Diffuse Large B-Cell Lymphoma |
732 |
of the CNS |
|
Immunodeficiency-Associated |
739 |
CNS Lymphomas |
|
AIDS-Related Diffuse Large B-Cell |
740 |
Lymphoma |
|
Lymphomatoid Granulomatosis |
741 |
Posttransplant |
743 |
Lymphoproliferative Disorder |
|
Intravascular (Angiocentric) |
745 |
Lymphoma |
|
Miscellaneous Rare CNS |
748 |
Lymphomas |
|
MALT Lymphoma of the Dura |
748 |
Metastatic Intracranial |
749 |
Lymphoma |
|
Histiocytic Tumors |
750 |
Langerhans Cell Histiocytosis |
751 |
Erdheim-Chester Disease |
754 |
Rosai-Dorfman Disease |
756 |
Juvenile Xanthogranuloma |
759 |
Histiocytic Sarcoma |
759 |
Hemophagocytic |
760 |
Lymphohistiocytosis |
|
Hematopoietic Tumors and Tumor- |
|
Like Lesions |
761 |
Leukemia |
761 |
Plasma Cell Tumors |
765 |
Extramedullary Hematopoiesis |
768 |
Lymphomas and Related
Disorders
Lymphoid CNS lesions may occur either as primary tumors or metastatic deposits from extracranial disease. Together, lymphoid neoplasms comprise the sixth most common group of CNS malignancies. More than 95% are diffuse large B-cell lymphomas (DLBCLs).
The remaining 4-5% of primary CNS lymphoma (PCNSL) subtypes are T- and NK-/T-cell lymphomas and low-grade B-cell lymphomas such as mucosaassociated (MALT) lymphomas of the dura and marginal zone B-cell lymphomas that correspond to their systemic counterparts. MALT lymphoma of the dura and Hodgkin-type lymphomas (HLs) are uncommon in

Neoplasms, Cysts, and Tumor-Like Lesions
732
(24-1) This is PCNSL. Periventricular lesions are in BG, thalamus, corpus callosum with extensive subependymal spread of disease .
(24-2) Autopsy shows PCNSL with bilateral deep basal ganglionic, thalamic masses , ependymal spread . (Courtesy R. Hewlett, MD.)
(24-3) Autopsy shows extensive ependymal spread of PCNSL around the lateral ventricles.(Courtesy R. Hewlett, MD.)
the CNS. Most are seen as metastatic lesions in the setting of advanced systemic disease although rare primary CNS lesions have been described.
We begin this chapter by focusing on DLBCL. We then consider the fascinating spectrum of immunodeficiency-associated CNS lymphomas
(including lymphomatoid granulomatosis and posttransplant lymphoproliferative disorder).
We follow the detailed discussion of DLBCL with a discussion of intravascular large B-cell lymphoma, an uncommon but increasingly wellrecognized cause of unexplained cognitive decline in elderly patients. Uncommon primary CNS lymphomas such as low-grade B-cell lymphomas, T- cell and NK-/T-cell lymphomas, anaplastic large cell lymphoma, and MALT lymphoma of the dura are then briefly discussed.
We close this section with a brief review of CNS metastatic lymphoma. (Metastatic lymphoma is included here instead of in Chapter 27 with other metastatic disease, as its imaging appearance differs from that of other systemic cancers that spread to the brain.)
Diffuse Large B-Cell Lymphoma of the CNS
Terminology
A diffuse large B-cell lymphoma of the CNS (DLBCL) is an extranodal B-cell lymphoma that arises exclusively inside the CNS. By definition, disease outside the nervous system is absent at the time of initial diagnosis. The vast majority of primary CNS lymphomas (PCNSLs) are DLBCLs. For purposes of discussion, in this text, the terms PCNSL and DLBCL are used interchangeably.
2016 WHO CLASSIFICATION OF PRIMARY CNS LYMPHOMAS
Diffuse Large B-Cell Lymphoma of the CNS
Immunodeficiency-Associated CNS Lymphomas
•AIDS-related diffuse large B-cell lymphoma
•Epstein-Barr virus-positive diffuse large B-cell lymphoma, NOS
•Lymphomatoid granulomatosis
•Posttransplant lymphoproliferative disorder
Intravascular Large B-Cell Lymphoma
Miscellaneous Rare CNS Lymphomas
•Low-grade B-cell lymphomas
•T-cell and NK-/T-cell lymphoma
•Anaplastic large cell lymphoma
MALT Lymphoma of the Dura
Etiology
General Concepts. Whether PCNSL arises inside or as transformed B cells originating outside the brain is unclear. Although functional lymphatic vessels are present in the dural venous sinuses, the brain parenchyma itself lacks classic lymphatics and normally contains very few lymphocytes. So how and why lymphomas arise as primary CNS neoplasms in immunocompetent individuals is unknown.
What is clearly evident is that lymphoma cells—regardless of whether they originate within or outside the brain—exhibit a distinct, highly selective neurotropism for the CNS microenvironment and its vasculature. Intraand perivascular tumor spread is common.
Lymphomas and Hematopoietic and Histiocytic Tumors
Most PCNSLs are mature B-cell lymphomas that have a late or postgerminal center of origin with blocked terminal B-cell differentiation. Persistent surface expression of both B-cell lymphoma 6 (BCL6) protein and interferon regulatory factor 4 (IRF4) along with immunoglobulin gene rearrangement indicates that most PCNSLs have an activated B-cell phenotype.
Genetics. No genetic predispositions to PCNSL have been identified. Next-generation sequencing has identified a number of mutated genes involved in B-cell proliferation and differentiation, but to date no true lymphomagenesis "driver mutations" similar to those of gliomagenesis have been pinpointed.
Over 80% of protein-changing mutations in CNS DLBCL are located in eight specific genes (including PTEN, CTNNB1, ATM, and TP53), pointing to the potential role of these genes in lymphomagenesis. Notably, CNS DLBCL shares certain gene mutations (e.g., TP53 and SMO) with other solid brain tumors.
Epigenetic changes may also contribute to DLBCL pathogenesis, including aberrant hypermethylation of DAPK1.
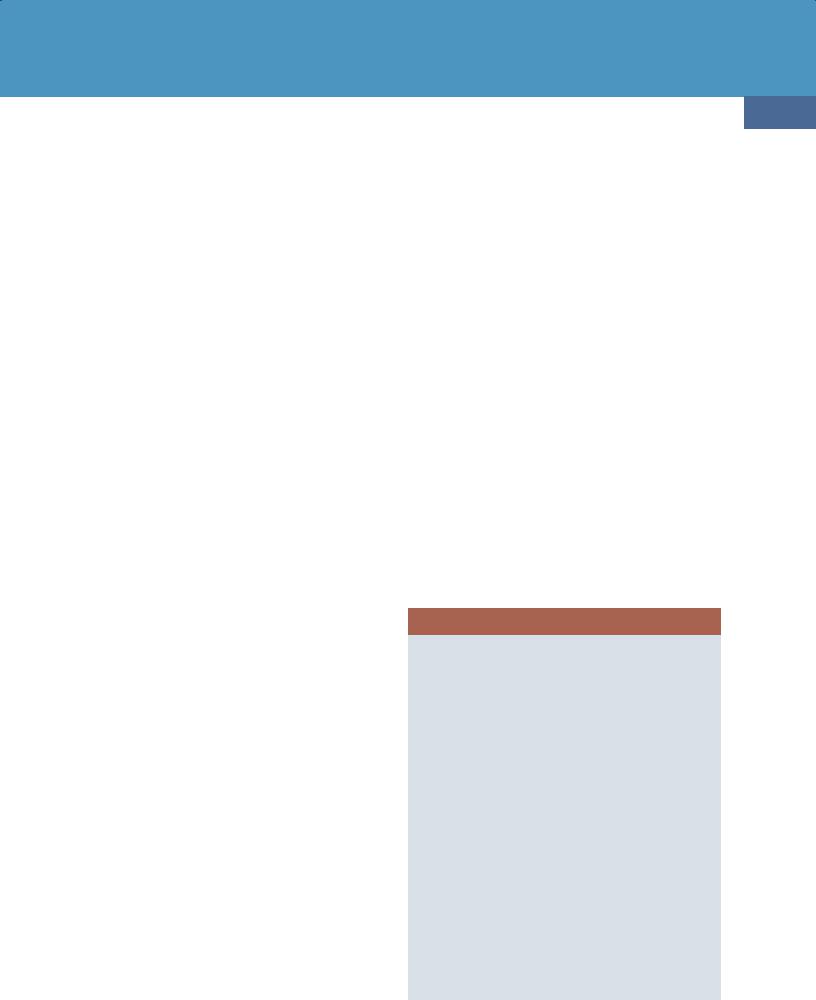
Pathology
Location. DLBCL can affect any part of the neuraxis. Up to 75% all PCNSLs contact a CSF surface, either the ventricular ependyma or pia (24-1). The cerebral hemispheres are the preferred site (85%). Lesions are often deep-seated with a predilection for the periventricular white matter, especially the corpus callosum. The basal ganglia and thalami are the next most common locations (24-2). Tumor spread along the ventricular ependyma and into the choroid plexus is seen in some cases (24-2) (24-3).
The hypothalamus, infundibulum, and pituitary gland are less common sites. Posterior fossa lesions and lesions of the spinal cord are relatively rare (15% of cases).
Primary DLBCL may also develop in the leptomeninges, calvarial vault, and central skull base although these areas are more commonly involved by metastatic spread from extracranial primary tumors (see below). Primary dura-based DLBCLs are very rare. Most are low-grade B-cell or MALT types.
Ocular lymphomas are almost always high-grade B-cell lymphomas. In contrast, lymphomas of the orbital adnexa are most often MALT-type tumors (see below).
Size and Number. DLBCL lesions vary in size from microscopic implants to large bulky masses.
Up to two-thirds of all PCNSLs are solitary lesions. Of the PCNSLs with multiple lesions, approximately half are bilateral.
Gross Pathology. Single or multiple hemispheric masses with a "fish flesh" consistency are typical. In contrast to astrocytomas, lymphomas tend to be relatively well demarcated rather than diffusely infiltrating lesions. Most are solid, pale lesions with occasional necroses and small hemorrhagic foci. Large confluent areas of frank necrosis and
733
gross intratumoral hemorrhage are more common in AIDSrelated PCNSLs (see below) (24-10).
Occasionally, PCNSLs diffusely infiltrate large areas of the hemispheres without forming a focal cohesive mass. Widespread infiltration of lymphoma cells in both gray and white matter is characteristic. This condition—also known as lymphomatosis cerebri—is uncommon, occurring in less than 5% of cases, and is a pattern, not a distinct disease entity.
Microscopic Features. PCNSLs are highly cellular tumors. Microscopically, large atypical cells with large round to irregular nuclei with prominent nucleoli are typical. MIB-1 is high, often exceeding 50% (significantly higher than with glioblastoma). The WHO does not assign a grade to PCNSL.
Most PCNSLs exhibit a distinct predilection for blood vessels, with layering of tumor cells in and around vessels that extend from these perivascular cuffs into the adjacent parenchyma. This "angiocentric" clustering is often accompanied by prominent rings of reticulin in and around vessel walls.
The overwhelming majority of DLBCLs are positive on CD20 and CD45 immunostaining.
Reactive inflammatory infiltrates consisting of mature T and B cells are common. Occasionally, steroid-responsive multifocal demyelinating "sentinel" lesions that are indistinguishable from those seen in tumefactive multiple sclerosis or acute disseminated encephalomyelitis (ADEM) can be seen months or even several years before PCNSL is diagnosed.
Even small doses of corticosteroids can induce striking apoptosis in PCNSLs. Steroid treatment preceding biopsy can obscure the diagnosis of PCNSL in up to 50% of cases!
PCNSL: ETIOLOGY AND PATHOLOGY
Etiology
•CNS lacks lymphatics, normally contains few lymphocytes
•Precise origin in immunocompetent individuals unknown
Pathology
•3-6% of all primary CNS neoplasms
•Vast majority (90-95%) are DLBCLs
○Large atypical cells
○CD20+, CD45+
○MIB-1 > 50%
•Predilection for deep brain
○Periventricular WM, basal ganglia
○Perivascular lymphoid clusters also common
•Solitary (2/3), multiple (1/3)
○Multiple compartments may be involved
•Focal > > diffusely infiltrating lesions (lymphomatosis cerebri)
•Hemorrhage, necrosis rare in immunocompetent patients
•"Sentinel" demyelinating lesion indistinguishable from MS or ADEM can precede PCNSL!
•Corticosteroids → ↑ apoptosis, can obscure diagnosis of PCNSL!

Neoplasms, Cysts, and Tumor-Like Lesions
734
Clinical Issues
Epidemiology. Although PCNSLs account for only 3% of all malignant CNS neoplasms, worldwide prevalence is increasing as a result of the HIV/AIDS epidemic and the use of immunosuppressive therapies (see below).
Demographics. Although PCNSLs can occur at all ages, they are generally tumors of middle-aged and older adults. Peak age (in immunocompetent patients) is 60 years. There is an overall slight male predominance.
Presentation. Most patients with PCNSL present with focal neurologic deficits, altered mental status, and neuropsychiatric disturbances. Seizures are less common than in patients with other primary brain tumors.
Occasionally, PCNSL is preceded by demyelinating and inflammatory lesions similar to multiple sclerosis. Cases of so-
called "sentinel lesions" occurring up to 2 years after initial presentation with demyelinating lesions have been reported.
Patients with nonfocal, diffusely infiltrating PCNSLs (lymphomatosis cerebri) typically present with generalized cognitive decline and rapidly progressive dementia. Memory deficits, ataxia, personality changes, and abnormal behaviors are common. Most patients are middle aged or elderly.
Natural History. CNS DLBCL has a significantly worse outcome than its systemic counterparts. CNS DLBCL is an aggressive tumor with a median survival of only a few months in untreated patients. In general, immunocompetent patients younger than 60 years fare slightly better than older patients and patients with acquired immunodeficiency syndromes.
Patients with lymphomatosis cerebri generally have a dire prognosis, with survival under 2 years uncommon.
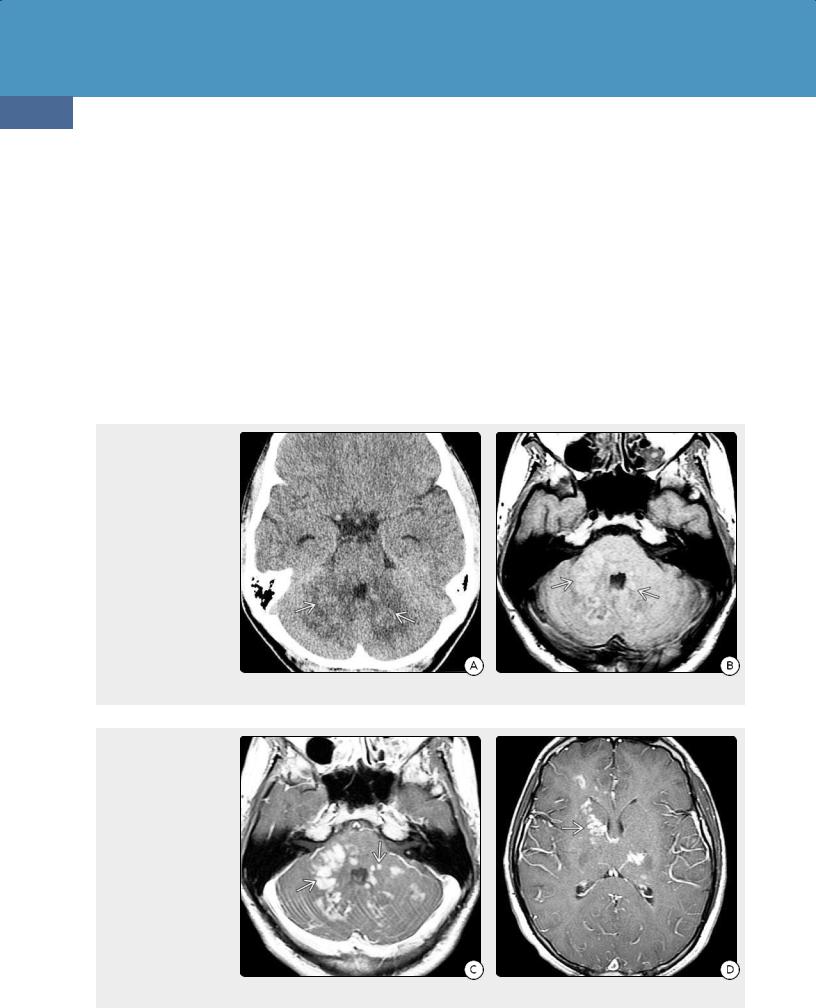
(24-4) NECT in a 63y woman with right-sided weakness shows a solitary hyperdense mass in the left BG with moderate edema . (24-5) Axial NECT in a 63y woman with "stroke-like" presentation shows a grossly thickened corpus callosum splenium and right occipital confluent hypodensity . Note that the "butterfly" mass is isodense with cortex and basal ganglia, not normal white matter. DLBCL was found at surgery.
(24-6A) NECT scan in a patient with PCNSL shows hyperdense masses in the left cerebellum . (Courtesy P. Hilenbrand, MD.) (24-6B) CECT shows multiple moderately enhancing masses in the cerebellum. (Courtesy P. Hildenbrand, MD.)
Lymphomas and Hematopoietic and Histiocytic Tumors
Treatment Options. Early diagnosis is crucial for proper management of PCNSLs. As gross surgical resection does not improve prognosis, stereotactic biopsy for diagnostic confirmation and histologic tumor typing is recommended.
As with systemic non-Hodgkin lymphomas, treatment options for DLBCL include corticosteroids, high-dose methotrexatebased polychemotherapy, and radiation. Approximately 70% of patients initially respond to treatment, but relapse is very common. Progression-free survival is approximately 1 year, and overall survival is approximately 3 years. Only 20-40% of patients experience prolonged progression-free survival.
Antineoplastic agents designed specifically to treat B-cell malignancies and B-cell-driven diseases such as rheumatoid arthritis have been used with some success in selected cases. Rituximab is a chimeric murine/human monoclonal immunoglobulin G1 antibody that targets CD20, a cell-surface marker specifically found on B lymphocytes.
735
Imaging
General Features. Contrast-enhanced cranial MR is the modality of choice in evaluating patients with suspected PCNSL. As isolated spinal cord involvement is rare (3-4% of cases), spinal imaging is indicated only in patients with myelopathy or suspected diffuse meningeal dissemination.
Contrast-enhanced CT of the chest, abdomen, and pelvis or PET-CT is generally recommended in patients with suspected PCNSL to look for an extracranial source of disease.
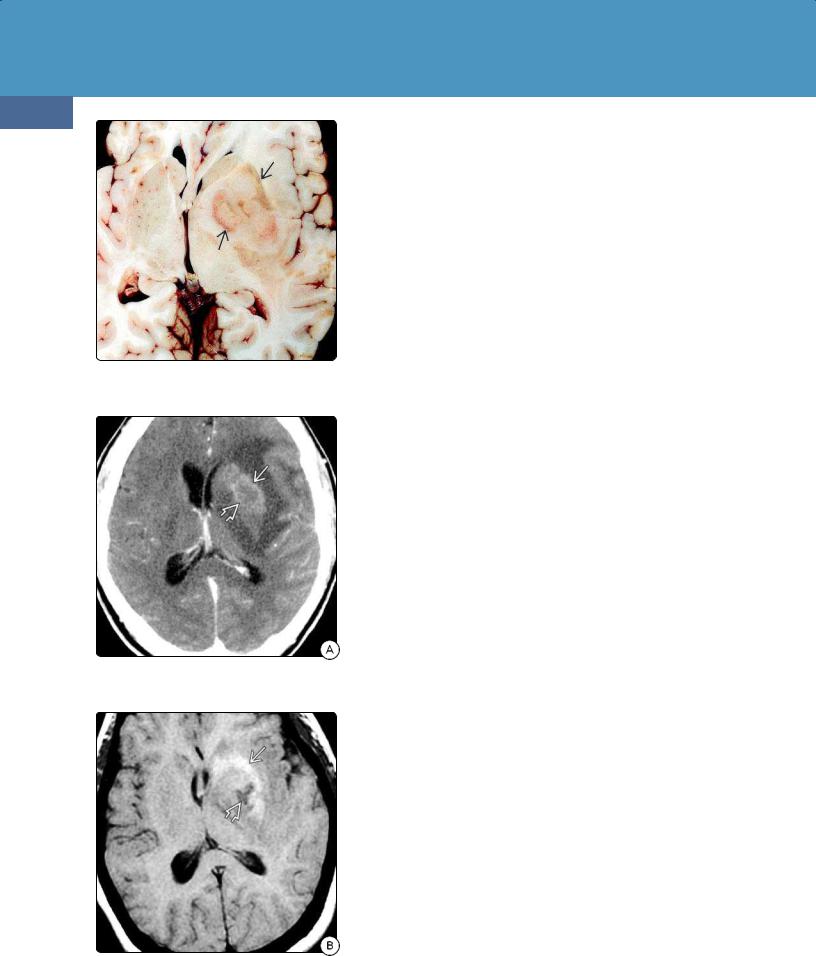
CT Findings. All PCNSLs are highly cellular tumors. White matter or basal ganglia lesions in contact with a CSF surface are typical. Most lesions appear hyperdense compared with normal brain on NECT scans (24-4) (24-5). Marked peritumoral edema is common, but gross necrosis, hemorrhage, and calcification are rare (2-5%) unless the patient is immunocompromised (24-11A).
(24-7A) Axial T1WI shows a mass in the left basal ganglia that is mostly isointense with cortex. (24-7B) T2WI in the same case shows that the massis mixed isoto hypointense relative to cortex. A moderate amount of hyperintense peripheral edema is present.
(24-7C) ADC map shows that the mass exhibits mild to moderate restricted diffusion . (24-7D) T1 C+ FS shows that the mass enhances intensely and uniformly. DLBCL was found at surgery (same case as Figure 24-4).
Neoplasms, Cysts, and Tumor-Like Lesions
736
(24-8A) Axial T1WI in a
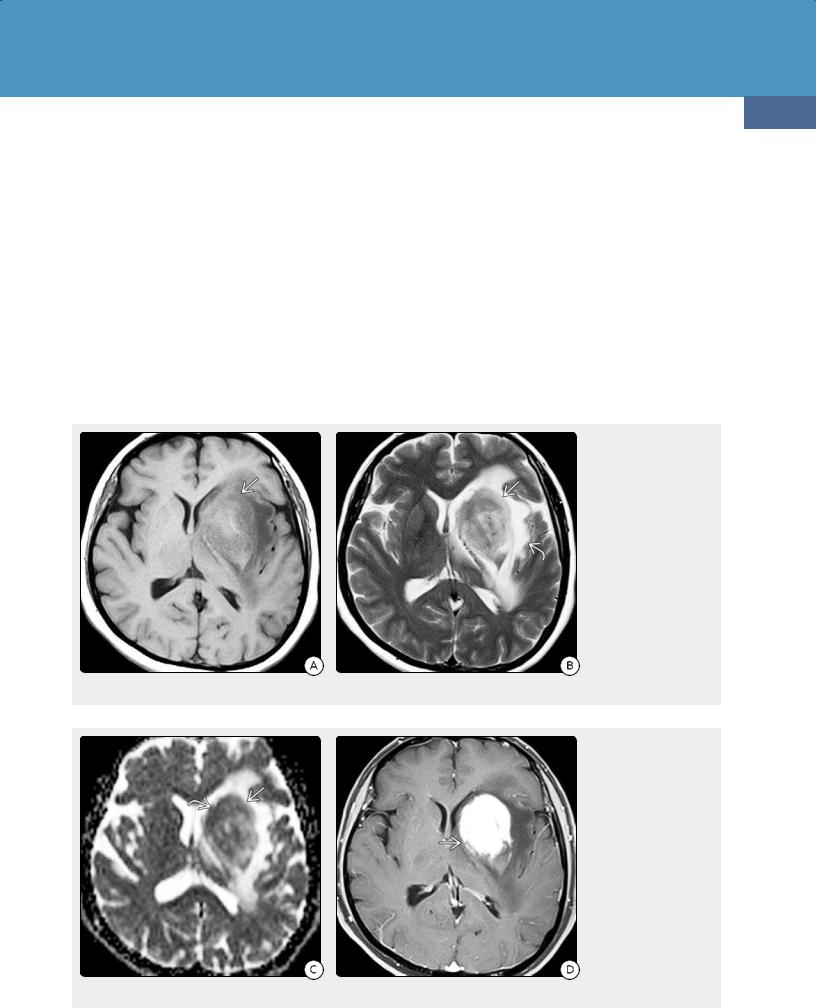
61y woman with DLBCL and no evidence for systemic disease on PET shows lesions in the basal ganglia and thalami that appear isointense with gray matter. (24-8B) T2WI in the same case shows that the lesions appear isointense with gray matter, associated with some hyperintense peripheral edema .
(24-8C) T1 C+ FS shows that the lesions enhance strongly and uniformly. (24-8D) The lesions show restricted diffusion with low ADC. Peritumoral edema is hyperintense because of "T2 shine-through."
(24-8E) pMR shows no evidence for elevated rCBV in the lesions .
(24-8F) pMR with Ktrans shows elevated capillary permeability in the lesions.
Lymphomas and Hematopoietic and Histiocytic Tumors
737
(24-9A) Axial T1WI in a
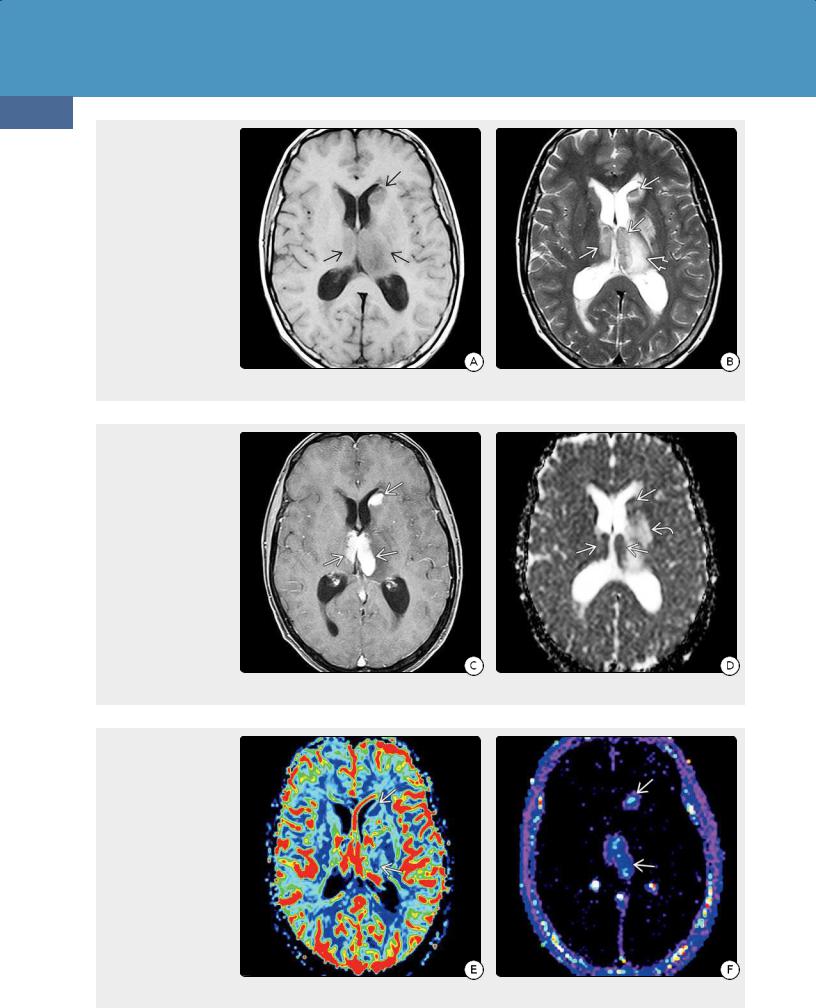
64y woman with altered mental status and increasing confusion shows enlargement and diffuse confluent hypointensity in both frontal lobes and the corpus callosum genu. (24-9B) Axial T2WI in the same case shows confluent hyperintensityand gyral expansion of both frontal lobes with maintenance of the underlying architecture of the brain.
(24-9C) Sagittal FLAIR in the same case shows infiltration and expansion of the corpus callosum . (24-9D) DWI in the same case shows restricted diffusion throughout the periventricular WM .
(24-9E) T1 C+ FS shows patchy enhancement in both frontal lobes. (24-9F) Coronal T1 C+ SPGR shows that the thickened corpus callosum is largely nonenhancing, while there is patchy punctate and linear enhancement in the periventricular and deep WM . Biopsy showed diffusely infiltrating DLBCL and lymphomatosis cerebri pattern.
Neoplasms, Cysts, and Tumor-Like Lesions
738
(24-10) Autopsy of PCNSL in HIV/AIDS shows hemorrhagic, necrotic left basal ganglia mass . (Courtesy R. Hewlett, MD.)
(24-11A) CECT scan shows necrosis and only faint rim enhancement in this HIV-positive patient with PCNSL.
(24-11B) T1WI shows T1 shortening due to subacute hemorrhage with more acute hemorrhage in the necrotic core of the lesion .
DLBCLs in immunocompetent patients show mild to moderate, relatively homogeneous enhancement on CECT (24-6). Irregular ring enhancement is rare unless the patient is immunocompromised.
MR Findings. Over three-quarters of DLBCLs in immunocompetent patients are isoor slightly hypointense compared with gray matter on T1WI and isointense on T2WI (24-7).
FLAIR signal is variable but usually isoor hyperintense. Microhemorrhages with intratumoral "blooming" on T2* are present in 5-8% of cases, but gross hemorrhage is uncommon unless the patient is immunocompromised (see below).
Nearly all PCNSLs in immunocompetent patients enhance (24-7D). Solid homogeneous or mildly heterogeneous enhancement is common; ring enhancement is rare. MRS typically demonstrates elevated choline and high lipids.
Because of their high cellularity, over 95% of PCNSLs show mild to moderate diffusion restriction with low ADC values (24-7C). MRS is nonspecific, with elevated choline, reduced NAA and myoinositol, and prominent lipids. Tumor neovascularization is absent in PCNSLs; therefore, rCBV is relatively low, and permeability is not increased on DCE pMR even though the tumor is highly malignant (24-8).
Diffusely infiltrating PCNSLs, also known as lymphomatosis cerebri, are uncommon. Patchy or confluent bihemispheric T2/FLAIR hyperintensities in the deep cerebral white matter and basal ganglia are typical (24-9). Enhancement may be minimal or even absent on initial imaging in lymphomatosis cerebri, but patchy or nodular enhancing lesions are common on follow-up studies. Restricted diffusion is seen in two-thirds of cases.
Steroid administration significantly alters the imaging findings of PCNSLs. Significant cell lysis with tumor regression and normalization of the bloodbrain barrier occurs in 40-85% of patients with corticosteroid-treated PCNSLs. Tumors typically diminish in size. Contrast enhancement also decreases or even disappears completely ("ghost tumor") although some T2 and FLAIR signal abnormalities may persist.
FDG PET Findings. Increased metabolism on FDG-PET is uncommon. However, occult systemic lymphomas are found in 5-8% of patients with putative PCNSL. In such cases, FDG PET and PET/CT fusion imaging are helpful in looking for extracranial lymphoma.
Differential Diagnosis
The major differential diagnosis of PCNSL is glioblastoma (GBM). Although both tumors often cross the corpus callosum, hemorrhage and necrosis are rare in PCNSL. Enhancement in immunocompetent patients with PCNSL is strong and relatively homogeneous, whereas a peripheral ring pattern is more typical of GBM. Advanced MR techniques such as DWI, MRS, and DCE pMR are helpful in distinguishing PCNSLs from other highly aggressive primary brain tumors.
The second most common differential diagnosis of PCNSL is metastasis. Dura-based PCNSLs may resemble meningioma or—due to their hyperdensity—even look like an acute epior subdural hematoma.
In patients with lymphomatosis cerebri who lack a dominant cohesive tumor mass, the differential diagnosis is much broader. Lymphomatosis cerebri can mimic infectious or autoimmune inflammatory encephalitis, microvascular disease, toxic-metabolic processes, and
Lymphomas and Hematopoietic and Histiocytic Tumors
neurodegenerative disorders. Callosal involvement favors lymphomatosis cerebri.
In the setting of solid organ or hematopoietic stem cell transplants, lymphomatoid granulomatosis and posttransplant lymphoproliferative disorder (PTLD) may closely resemble PCNSL. Biopsy is necessary for confirmation and patient management.
PCNSL: IMAGING AND DDx IN IMMUNOCOMPETENT PATIENTS
General Features
•Periventricular WM, basal ganglia common sites
○95% contact a CSF surface
•Cautions
○Findings vary with immune status
○Steroids may mask/↓ imaging findings!
CT
•Hyperdense on NECT
•Hemorrhage, necrosis rare
MR
•Generally isointense with GM on T1-, T2WI
•Petechial hemorrhage in immunocompetent patients
•Gross hemorrhage, necrosis rare
•Strong, relatively uniform enhancement
•Often restricts on DWI
•Lymphomatosis cerebri
○Mimics diffuse WM disease with T2/FLAIR confluent hyperintensity
○Enhancement can be subtle or patchy, occasionally absent
Differential Diagnosis
•Glioblastoma, metastasis
•Lymphomatosis cerebri
○Microvascular disease
○Encephalitis (infectious, inflammatory, autoimmune)
○Toxic-metabolic disorders
○Diffusely infiltrating glioma
Immunodeficiency-Associated CNS
Lymphomas
Lymphomas associated with either inherited or acquired immunodeficiency are grouped together as immunodeficiency-associated CNS lymphomas. The immunodeficiency-associated CNS lymphomas include AIDS-related diffuse large B-cell lymphoma, Epstein-Barr virus (EBV)-positive DLBCL in elderly patients with no known immunodeficiency, lymphomatoid granulomatosis, and monomorphic or polymorphic posttransplant lymphoproliferative disorders.
Some lymphomas are linked to viral infections. PCNSLs associated with EBV account for 10-15% of all cases.
Congenital immunodeficiency syndromes increase the risk of lymphoma, as does severe acquired immunosuppression. Congenital immunodeficiency syndromes include Wiskott-Aldrich syndrome and severe combined immunodeficiency.
Autoimmune diseases linked to PCNSL include rheumatoid arthritis, Sjögren syndrome, and systemic lupus erythematosus.
739
(24-12A) FLAIR in a 43y HIV+ man with ataxia shows a solitary right cerebellar lesion with surrounding edema , mass effect.
(24-12B) T1 C+ FS in the same case shows a thick, irregular enhancing rim surrounding a core of necrotic nonenhancing tissue .
(24-12C) DWI shows diffusion restriction in the rim and lesion core . Immunodeficiencyrelated DLBCL was found at surgery.
Neoplasms, Cysts, and Tumor-Like Lesions
740
AIDS-Related Diffuse Large B-Cell Lymphoma
Although HIV-associated PCNSL has become rarer with the introduction of highly active antiretroviral therapy (HAART), between 2 and 12% of HIV/AIDS patients eventually develop CNS lymphoma, generally during the later stages of their disease. These patients can exhibit elevated levels of B-cell- stimulatory cytokines and other markers of immune activation several years prior to the diagnosis of systemic AIDSassociated non-Hodgkin B-cell lymphomas.
Pathology
With a few exceptions, AIDS-related CNS DLBCL shares the morphological features of PCNSL in immunocompetent patients. In addition to EBV association, multiple lesions are more common, as are larger, more confluent necrotic areas
(24-10).
(24-13A) NECT scan in a
16y boy with fever, cough, and ataxia shows heterogeneous hypo-, isodense infiltrate in the white matter of both cerebellar hemispheres. (24-13B) Axial T1WI in the same patient shows that the multifocal cerebellar lesions are slightly hyperintense .
(24-13C) T1 C+ shows strong enhancement of the lesions . (24-13D) More cephalad T1 C+ scan shows additional enhancing foci in the basal ganglia and hemispheric white matter. Imaging diagnosis was hemophagocytic lymphohistiocytosis. Biopsy disclosed lymphomatoid granulomatosis.
Clinical Issues
Mean age at onset in patients with HIV/AIDS is 40 years, two decades younger than immunocompetent patients. Lymphomas in transplant recipients occur even earlier, generally between the ages of 35 and 40. Mean age of onset in children with inherited immunodeficiencies is 10 years.
Overall survival in HIV/AIDS and other immunocompromised patients with PCNSL is substantially diminished, no matter the age at presentation.
Imaging
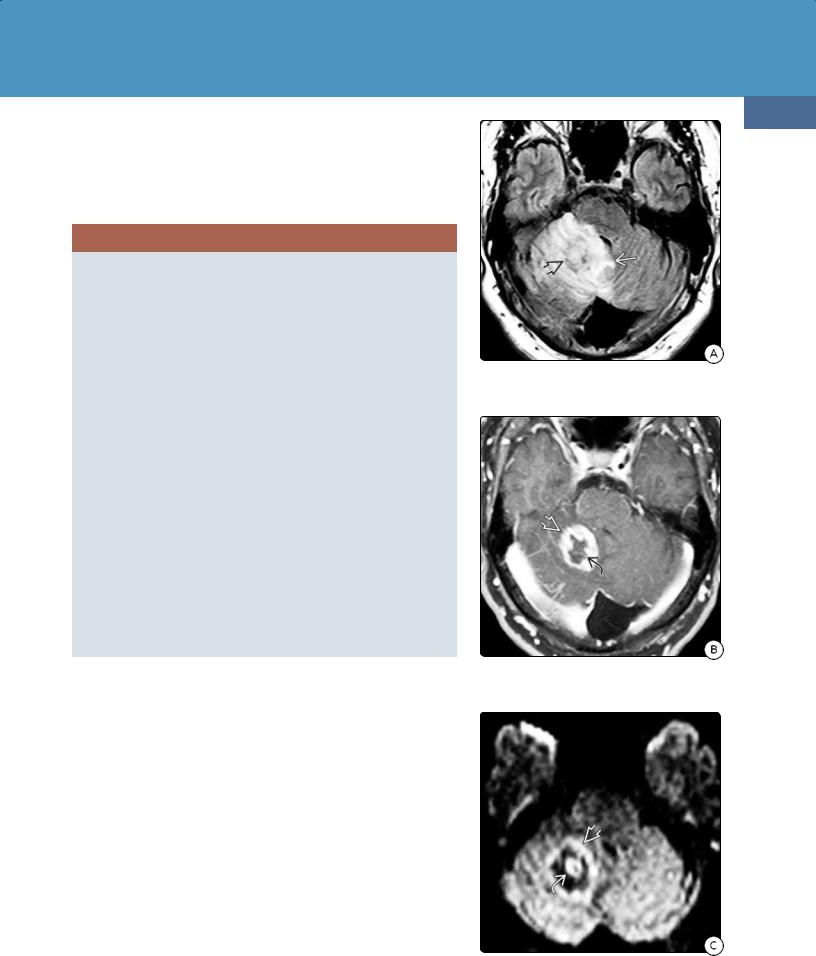
Multiple lesions are common, as are more frequent and larger confluent areas of necrosis (24-11A). Intratumor hemorrhage with T1 shortening and "blooming" on T2* scans is common (24-11B). Enhancement is variable but often mild. Ring enhancement surrounding a nonenhancing core of necrotic tissue is typical (24-12).

Lymphomas and Hematopoietic and Histiocytic Tumors
Differential Diagnosis
In immunocompromised patients, the major differential diagnosis of PCNSL is toxoplasmosis. A solitary ring-enhancing lesion in an HIV/AIDS patient is most often lymphoma, whereas multiple lesions are more characteristic of toxoplasmosis. An "eccentric target" sign is suggestive of toxoplasmosis although necrotic lymphomas occasionally show an enhancing "ring with a nodule" pattern. Toxoplasmosis is hypometabolic on PET and pMR and often exhibits elevated ADC values with a ratio greater than 1:6.
Progressive multifocal leukoencephalopathy (PML) usually does not enhance. However, acute PML lesions and JC virusassociated immune reconstitution inflammatory syndrome
(IRIS) may show ring enhancement. Enhancement often looks quite bizarre with multifocal poorly delineated partial rings of enhancement surrounding the demyelinating foci.
741
Lymphomatoid Granulomatosis
Terminology
Lymphomatoid granulomatosis (LYG) is a rare multisystem angiocentric and angioinvasive lymphoproliferative disorder characterized by atypical B-cell proliferations. LYG is now considered an immunodeficiency-related extranodal large B- cell lymphoma.
Etiology
EBV infection is a feature of most reported cases. LYG also occurs in the setting of HIV/AIDS and in patients on maintenance immunosuppression following solid organ transplantation. Inherited disorders such as Wiskott-Aldrich disease and congenital immunodeficiency syndrome are rare but reported causes of LYG.
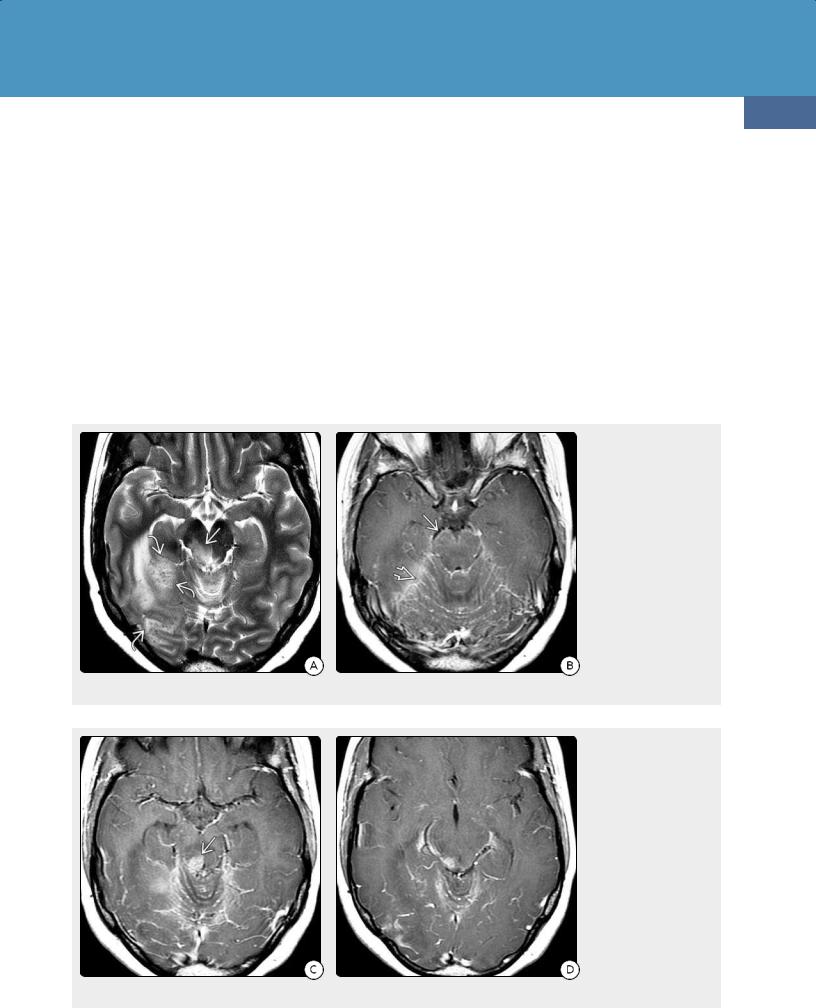
(24-14A) Axial T1 C+ FS in a 37y woman shows an enhancing mass in the right cerebellum and choroid plexus in 4th ventricle lateral recess . A less conspicuous mass is present in the left foramen of Luschka . (24-14B) More cephalad T1 C+ FS in the same case shows additional enhancing masses in the glomus of the left choroid plexus and right frontal dura-arachnoid. This is biopsy-proven lymphomatoid granulomatosis.
(24-15A) Axial T2WI in a
25y man with a grand mal seizure shows a mixed signal mass in the left parietal lobe. (24-15B) T1 C+ in the same case shows a ring-enhancing lesion. DWI (not shown) did not demonstrate restricted diffusion. This is biopsy-proven EpsteinBarr virus (EBV)-related lymphomatoid granulomatosis.

Neoplasms, Cysts, and Tumor-Like Lesions
742
Pathology
The lung is the most commonly involved site, followed by the skin. The CNS is involved in about 25% of cases. Lesions range in size from a few millimeters up to 1 or 2 cm in diameter.
Large focal masses are rare.
The classic histologic triad of LYG consists of nodular polymorphic lymphocytic infiltrate, angiitis, and granulomatosis with central necrosis.
LYG shares several histologic similarities with posttransplantation lymphoproliferative disorders (see below). Angiocentric and angiodestructive polymorphous infiltrates consisting predominantly of lymphocytes mixed with plasma cells, immunoblasts, and histiocytes are present.
LYG is graded on a scale of 1 to 3 based on the numbers of atypical EBV-positive B cells and necrosis compared with the background of reactive T lymphocytes.
(24-16A) T2WI in a 66y woman with altered mental status 3 years after orthotopic liver transplant shows multiple heterogeneously hyperintense lesions and severe left hemisphere edema. (Courtesy P. Chapman, MD.) (24-16B) T1 C+ MR in the same case shows multiple solid , ringenhancing lesions . (Courtesy P. Chapman, MD.)
(24-16C) Delayed cephalad T1 C+ FS scan shows multiple enhancing lesions . (Courtesy P. Chapman, MD.) (24-16D) Fused PET-contrast- enhanced MR image shows no evidence for increased metabolic activity in the lesions. Brain biopsy showed EBV+ PTLD. (Courtesy P. Chapman, MD.)
Clinical Issues
Patients of all ages are affected, but peak occurrence is between the fourth and sixth decades. The typical patient is a middle-aged man with fever, dry cough, and weight loss. As clinical manifestations, laboratory data, and imaging are all nonspecific, biopsy is mandatory for definitive diagnosis.
Prognosis is generally poor in untreated patients with median survival of 14 months. Approximately half of patients with LYG respond to steroids and chemotherapeutic agents.
Imaging
Nearly half of patients with LYG have demonstrable brain lesions on MR. Imaging findings are nonspecific, and there is no direct correlation with LYG grade.
The most common abnormalities are multifocal T2/FLAIR nodular hyperintensities in the cerebral hemispheres, deep
Lymphomas and Hematopoietic and Histiocytic Tumors
gray nuclei, cerebellum, or spinal cord (24-13). The second most common imaging finding in LYG is involvement of the leptomeninges and cranial nerves. Dura-based masses and choroid plexus lesions also occur (24-14).
LYG typically enhances strongly. Both solid and ring-like patterns as well as multifocal punctate and nodular and linear enhancing foci have been described (24-15).
Differential Diagnosis
The major differential diagnosis of LYG is vasculitis. Other considerations include diffusely infiltrating B-cell lymphoma
(lymphomatosis cerebri), intravascular lymphoma, and nonneoplastic lymphoproliferative disorders such as CLIPPERS (chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids).
743
Posttransplant Lymphoproliferative Disorder
Posttransplant lymphoproliferative disorder (PTLD) is a potentially life-threatening complication of immunosuppressive therapy in patients with solid organ or hematopoietic cell transplantation.
The PTLD disease spectrum ranges from an infectious mononucleosis-like illness with reactive lymph node hyperplasia to malignant lymphoma. PTLDs are related to EBV.
Pathology
PTLDs can be polymorphic or monomorphic. Polymorphic PTLD consists of a heterogeneous cell population that may reflect the full range of B-cell maturation from prominent plasmacytic infiltrates to proliferating immunoblasts with
(24-17A) Axial T2WI in a
28y woman who was immunosuppressed for rheumatoid arthritis shows multifocal confluent hyperintense lesions in the midbrain and right occipital lobe. (24-17B) T1 C+ in the same case shows diffuse linear and nodular enhancement along the midbrain and cerebellum .
(24-17C) More cephalad
T1 C+ shows an enhancing mass in the midbrain as well as the linear and nodular cerebellar and occipital enhancement. (24-17D) More cephalad T1 C+ shows nodular, sulcal, mass-like enhancement along the midbrain, superior vermis, and right occipital lobe. This was biopsy-proven PTLD.

Neoplasms, Cysts, and Tumor-Like Lesions
744
(24-18) Graphic depicts IVL. Malignant cells plug vessels, causing perivascular infiltrates and petechial hemorrhages.
(24-19) Autopsied IVL shows punctate/linear grayish infiltrates , petechial and perivascular hemorrhages . (Courtesy R. Hewlett, MD.)
(24-20) Intravascular lymphoma completely fills brain arteriole with "small round blue cells." (Courtesy T. Tihan, MD.)
necrosis. Polymorphic PTLD does not fulfill the histologic criteria for lymphoma.
Monomorphic PTLD consists of large blastic cells with prominent nucleoli. Most monomorphic PTLDs are classified as diffuse large B-cell lymphomas.
Clinical Issues
Epidemiology. The overall prevalence of PTLD following solid organ transplantation is 0.5-2.5%. Up to 15-20% of patients who develop PTLD have CNS involvement. Isolated CNS PTLD is rare, occurring in less than 0.5% of transplant cases.
Demographics. Pediatric transplant recipients develop PTLD more frequently than adult patients because children are less likely to have EBVspecific immunity at transplantation. The frequency varies with the type of transplant, with the highest prevalence reported with multiorgan or intestinal (20%), lung or heart (8-20%), liver (4-15%), and kidney (1-8%) transplants. PTLD after hematopoietic cell transplantation accounts for less than 2% of cases.
Presentation. PTLD typically presents several years following transplantation. Symptoms very with tumor location.
Treatment Options. Because treatment regimens vary significantly, distinguishing polymorphic from monomorphic PTLD and malignant lymphoma is essential, often requiring biopsy for definitive diagnosis.
Reduction or cessation of immunosuppression is generally the first step in treating PTLD. Polymorphic PTLD usually responds well within 2 to 4 weeks. PTLD that fails to respond to reduced immunosuppression has a very high mortality rate (50-90%).
Most cases of monomorphic PTLD are unresponsive to immunosuppression withdrawal and require additional therapeutic modalities. Surgical resection, chemotherapy, and radiation therapy are possibilities.
Because CNS PTLD lesions are often resistant to the combination of drugs known as CHOP (cyclophosphamide, hydroxydaunomycin, Oncovin, and prednisone), radiotherapy is generally used in combination with agents such as high-dose methotrexate. High-dose rituximab, an anti-CD20 monoclonal antibody, may be effective in some patients.
Imaging
Imaging features of CNS PTLD resemble those of AIDS-related lymphoma. Most lesions are solitary masses that are hypointense to cortex on T1WI and heterogeneously hypo-/hyperintense on T2WI. Solid or ring-like enhancement is common following contrast administration (24-16) (24-17). Lymphoma-related PTLDs usually show moderate restriction on DWI.
Extracranial head and neck PTLD results in a spectrum of findings. Bilateral cervical lymphadenopathy occurs in 75% of cases. Nodes often appear necrotic. Other manifestations include orbital involvement and sinonasal lesions that resemble polyposis or sinusitis.
Differential Diagnosis
The major differential diagnosis of PTLD is primary CNS lymphoma—especially AIDS-related lymphoma. The imaging differential diagnosis of CNS lesions in transplant recipients also includes opportunistic infections such as toxoplasmosis.
Lymphomas and Hematopoietic and Histiocytic Tumors
POSTTRANSPLANT LYMPHOPROLIFERATIVE DISORDER (PTLD)
Terminology and Etiology
•Complication of organ or stem cell transplants
○Long-term maintenance immunosuppression
•Ranges from benign mono-like illness to malignant lymphoma
Pathology
•Monomorphic or polymorphic lymphoid proliferations
•Polymorphic PTLD has plasmacytic B-cell elements of varying maturity
•Monomorphic PTLD has blastic cells → large B-cell lymphoma
Clinical Issues
•0.5-2.5% of patients with solid organ transplants
•15-20% with PTLD have CNS involvement
•Presents several years after transplantation
•Children > adults
Imaging
•Brain findings resemble those of AIDS-related lymphoma
•Extracranial PTLD
○Bilateral cervical adenopathy common
○Orbital, sinonasal lesions
Intravascular (Angiocentric) Lymphoma
Intravascular lymphoma (IVL) is a rare subtype of DLBCL characterized by proliferating malignant cells within small and medium-sized vessels. Although it can involve any organ, IVL typically affects the skin and the CNS.
Terminology
IVL is also called angiocentric or angioendotheliotropic lymphoma, angiotropic large cell lymphoma, endovascular lymphoma, and malignant angioendotheliomatosis.
Etiology
IVL is an aggressive malignant lymphoma that usually arises from B cells. T cells or NK cells may occasionally be the cell of origin. A possible association of IVL (especially the NK type) with EBV has been reported.
Pathology
The gross macroscopic appearance varies from normal to small multifocal infarcts of varying ages scattered throughout the cortex and subcortical white matter (24-18). Focal cerebral masses are rare. Petechial microhemorrhages may be present and are more common than confluent macroscopic bleeds (24-19).
At histologic examination, markedly atypical cells with large round nuclei and prominent nuclei are found in small and medium-sized vessels (24-20). Extension into the adjacent perivascular spaces is minimal or absent. CD20 staining is helpful in identifying tumor cells, especially when they are sparse and widely scattered.
Clinical Issues
Epidemiology. IVL is rare. CNS involvement occurs in 75-85% of patients.
Demographics. IVL is typically a tumor of middle-aged and elderly patients. Mean age at presentation is 60-65 years.
745
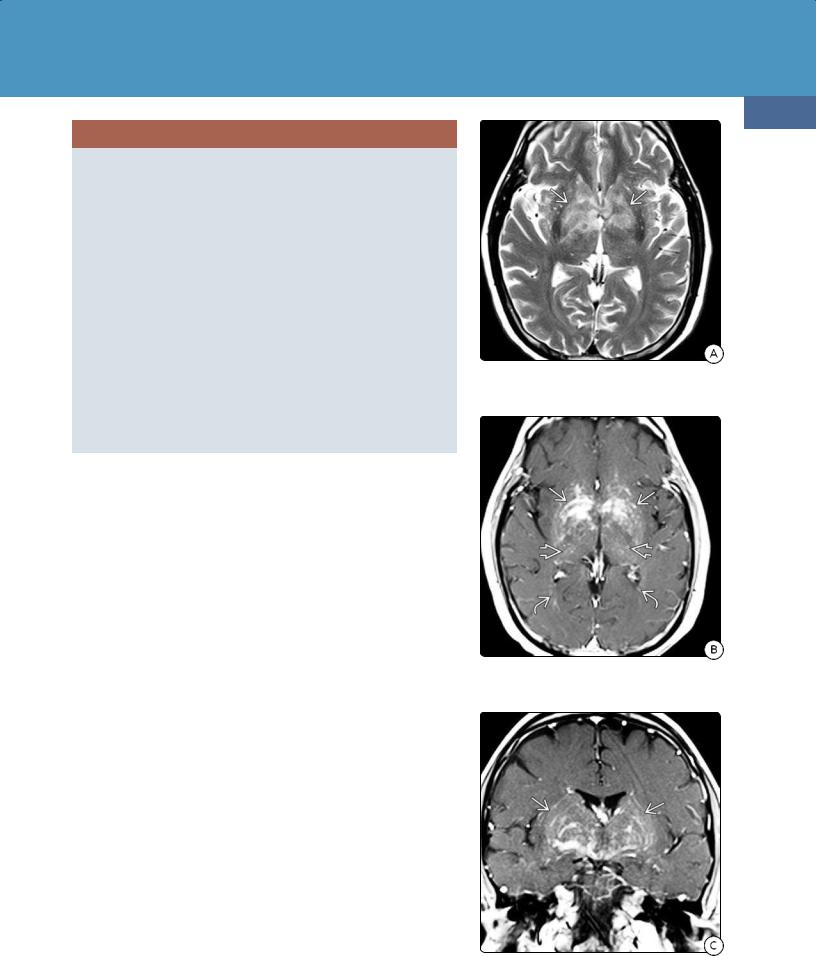
(24-21A) T2WI in a 61y man with progressive cognitive decline shows patchy hyperintensities in the basal ganglia and internal capsules .
(24-21B) Axial T1 C+ shows punctate, linear, and patchy confluent foci of contrast enhancement in the basal ganglia , thalami , deep WM .
(24-21C) Coronal T1 C+ shows prominent curvilinear enhancement following penetrating arteries , intravascular DLBCL found at biopsy.
Neoplasms, Cysts, and Tumor-Like Lesions
746
Presentation. Sensory and motor deficits, neuropathies, and multiple stroke-like episodes are common symptoms. Some patients present with progressive neurological deterioration and cognitive decline characterized by confusion and memory loss. Skin changes with elevated plaques or nodules are present in half of all cases.
Natural History. Outcome is generally poor. By the time of initial presentation, most patients have advanced disseminated disease. IVL is a relentless, rapidly progressive disease with a high mortality rate. Mean survival is 7-12 months.
Treatment Options. Because IVL is a widely disseminated disease, systemic chemotherapy is the recommended treatment. High-dose chemotherapy with autologous stem cell transplantation is often used in younger patients.
Imaging
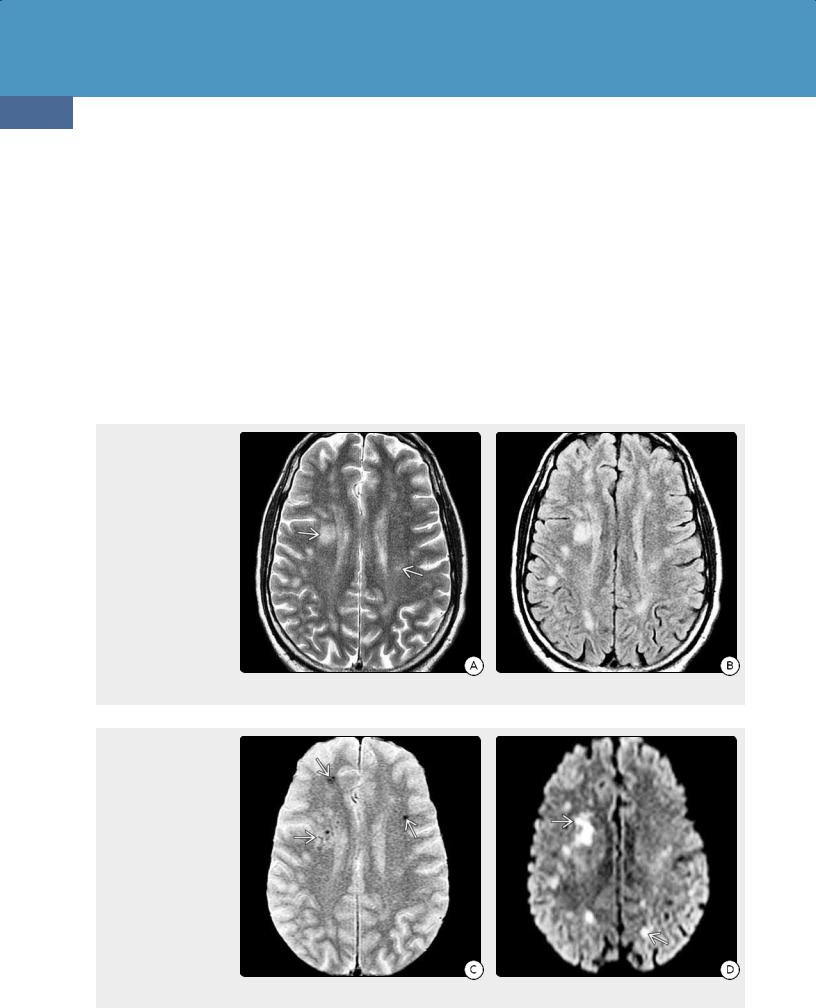
There are no pathognomonic neuroimaging findings for IVL. Ischemic foci with infarct-like lesions are the most common imaging finding. CT may be normal or nonspecific, demonstrating only scattered white matter hypodensities. MR shows multiple T2/FLAIR hyperintensities (24-21A). Microhemorrhages are common, so "blooming" foci on T2* (GRE, SWI) are often present (24-22). Linear/punctate enhancement oriented along perivascular spaces is suggestive of IVL (24-21C). Multifocal areas of diffusion restriction may be present (24-22D).
Differential Diagnosis
IVL is a "great imitator," both clinically and on imaging studies. Stereotactic biopsy is thus necessary to establish the definitive diagnosis. Vasculitis with punctate and linear enhancing foci
(24-22A) T2WI in a 60y man with a several-month history of increasing confusion and decreasing mental status shows multifocal ill-defined hyperintensities in the hemispheric WM . (2422B) FLAIR scan in the same patient shows several additional lesions. There was no evidence of cortical or basal ganglia hyperintensities.
(24-22C) T2* GRE shows multiple tiny hemorrhagic foci . (24-22D) DWI shows multiple foci of restricted diffusion in both hemispheres . The patient died shortly after the scan was obtained. Autopsy demonstrated intravascular lymphoma.
Lymphomas and Hematopoietic and Histiocytic Tumors
may be virtually indistinguishable from IVL on imaging studies alone.
PCNSL, especially in the setting of immunodeficiency syndromes, may mimic IVL. IVL is most often multifocal, whereas two-thirds of PCNSLs are solitary lesions. Diffuse multifocal PCNSL, especially when it occurs in the form of lymphomatosis cerebri, may be difficult to distinguish from IVL. Lymphomatosis cerebri often shows little or no enhancement.
Rapidly progressive leukoencephalopathy with confluent nonenhancing white matter lesions is a rare presentation of diffusely infiltrating CNS IVL and may mimic a cerebral demyelinating disorder.
Diffuse subacute viral encephalitis can likewise mimic IVL, especially on biopsy. Parenchymal neurosarcoid with perivascular nodular spread may also resemble IVL on imaging studies.
747
INTRAVASCULAR (ANGIOCENTRIC) LYMPHOMA
Pathology
•Small/medium-sized vessels filled with tumor
•Little/no parenchymal tumor
•Multifocal infarcts, microhemorrhages common
Clinical Issues
•Older patients with dementia, cognitive decline, TIAs
•Skin lesions (50%)
Imaging
•Multifocal T2/FLAIR hyperintensities
•Hemorrhages, foci of restricted diffusion
•Linear/punctate enhancement
Common Differential Diagnoses
•PCNSL
•Vasculitis
(24-23A) T2WI in a 52y woman with PCNSL, diffuse large T-cell type, shows hyper-, hypointense mass with striking peritumoral edema .
(24-23B) The mass restricted on DWI (not shown). T1 C+ scan shows that the mass enhances strongly and relatively uniformly.
(24-24A) Axial T2 FS scan in a 26y man with EBVpositive extranodal NK-/T- cell lymphoma shows a hypointense mass invading the upper nasopharynx , skull base, and cavernous sinuses. (24-24B) Sagittal T1 C+ in the same case shows the enhancing nasopharyngeal mass invading the skull base, extending intracranially to involve the pituitary gland and stalk . Note dural mass along the clivus .
Neoplasms, Cysts, and Tumor-Like Lesions
748
Miscellaneous Rare CNS Lymphomas
Other than DLBCL, primary CNS lymphomas are rare. These include low-grade B- and T-cell lymphomas, high-grade T- and NK-/T-cell lymphomas (NKTCLs), and the exceptionally rare primary CNS Hodgkin and Burkitt lymphomas.
Low-grade lymphomas, most of which are of B-cell lineage, account for approximately 3% of all CNS lymphomas and almost exclusively affect immunocompetent adults. These tend to be indolent neoplasms with relatively longer survival compared with DLBCLs.
Primary CNS T-cell lymphomas and NKTCLs are very rare, accounting for approximately 2% of all PCNSLs. Most reported cases are associated with prior EBV infection. Imaging findings are generally indistinguishable from parenchymal DLBCL, with solitary, multifocal, or diffusely infiltrating masses (24-23). Rare cases of primary NKTCLs presenting as diffuse leptomeningeal and cranial nerve
(24-25A) Coronal T1 C+ in a 73y woman with forehead swelling and blurry vision in her left eye shows orbital adnexal and dural-based masses. (24-25B) T1 C+ FS of the brain shows diffuse bilateral dura-arachnoid thickening and focal calvarial masses . Extranodal marginal zone lymphoma of mucosaassociated lymphoid tissue (MALT lymphoma) of the dura was found at surgery.
(24-26A) Axial T1 C+ FS in a 67y woman with blurry vision and right CN VI palsy shows a dural-based infiltrating mass in the right cranial fossa , cavernous sinus . (2426B) Coronal T1 C+ FS in the same case shows the right cavernous sinus , middle fossa duraarachnoid , and temporalis muscle thickening . This was MALT lymphoma of the dura.
enhancement have been reported. Approximately 7% of patients with peripheral T-cell lymphoma develop secondary CNS disease.
EBV-positive nasal-type extranodal NKTCL primarily affects young to middle-aged adult men. The upper aerodigestive tract (nasal cavity, nasopharynx, paranasal sinuses, and palate) is most commonly involved. Preferential sites of extranasal involvement include the skin, soft tissue, GI tract, and testis. Less than 3% of cases invade or metastasize to the CNS (2424). Prognosis is generally poor even with chemotherapy and radiation therapy.
MALT Lymphoma of the Dura
Extranodal marginal zone lymphomas of mucosaassociated lymphoid tissue (MALT lymphomas) in the head and neck are most often ocular adnexal tumors, occurring in the conjunctiva, lacrimal glands, orbit, and eyelids. The cranial meninges, especially the dura, are occasional intracranial sites

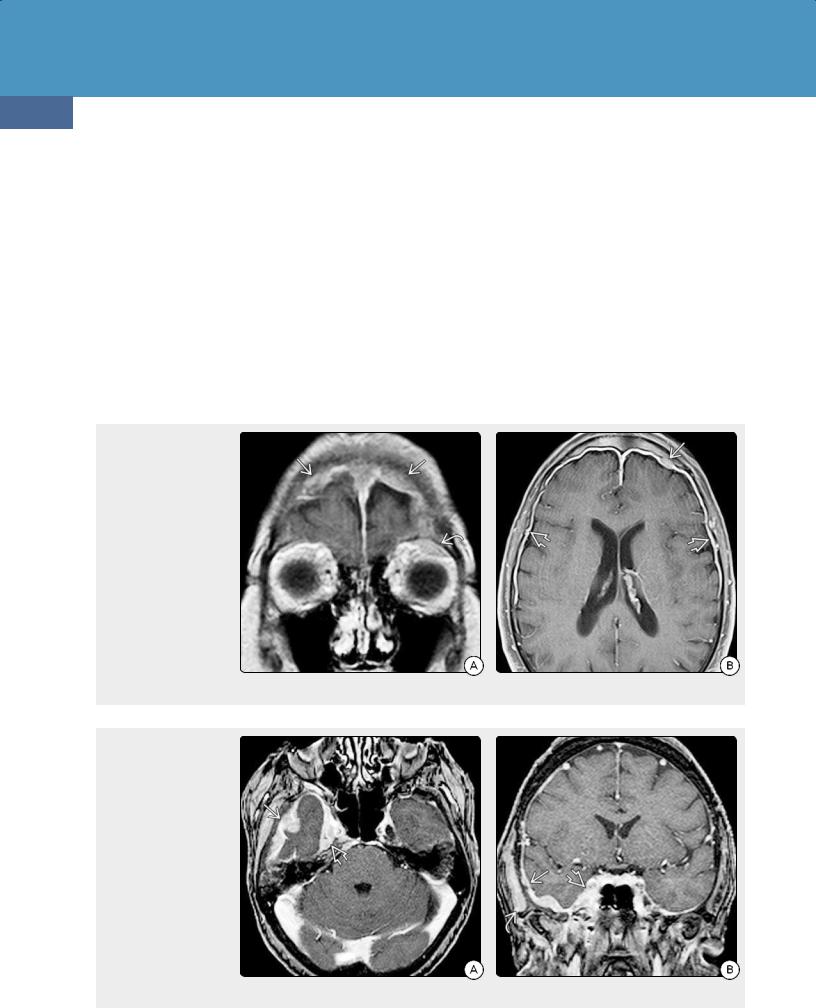
Lymphomas and Hematopoietic and Histiocytic Tumors
(24-25). Diffuse dura-arachnoid thickening with one or more meningioma-like masses is the most typical imaging finding (24-26). Parenchymal lesions are rare. Prognosis is excellent, with a 5-year survival rate of 85%.
Metastatic Intracranial Lymphoma
Terminology
Metastatic intracranial lymphoma is also called secondary CNS lymphoma (SCNSL). Here the skull, meningeal, and brain lesions are all secondary to systemic lymphoma (24-27) (2428) (24-29) (24-30).
Clinical Issues
Aggressive high-grade tumors increase the risk of CNS spread. Between 3-5% of patients with diffuse large B-cell systemic lymphomas eventually develop CNS involvement. Approximately 80% of SCNSLs are caused by DLBCL.
749
Peak prevalence of metastatic CNS lymphoma is in the sixth and seventh decades. Prognosis is poor, especially in elderly patients. Systemic methotrexate has been recommended as the optimal treatment for isolated CNS relapse involving the brain parenchyma. Rituximab treatment may be effective in some cases.
Imaging
SCNSL is typically identified with neuroimaging. Metastatic lymphoma rarely presents as a parenchymal mass. In contrast with PCNSL, skull and dural involvement is much more frequent (24-31) (24-32). Both calvarial vault and skull base metastases are common. Skull base metastases may extend inferiorly into the nose and paranasal sinuses or spread superolaterally into the cavernous sinus, Meckel cave, and pituitary gland/stalk.
Calvarial lesions often involve the adjacent scalp and epidural space. Dural "tails" are common. Involvement of the
(24-27) Close-up view shows leptomeningeal metastases . Metastatic lymphoma also thickens the infundibular stalk . The pituitary gland (not shown) was also involved by tumor. (24-28) Nodular, linear metastases from systemic lymphoma diffusely thicken the nerve roots of the cauda equina . Axial section shows that the intradural, extramedullary space is filled with tumor .
(24-29) Cut section through the ventricles shows that the ependymal surfaces of the lateral ventricles are coated with glistening tumor . The perivascular spaces in the basal ganglia are enlarged by cords of intravascular malignant lymphoma . (24-30) Diffuse dural thickeningwith multiple focal deposits of lymphoma resembles meningiomatosis. (Autopsy cases are courtesy R. Hewlett, MD.)