

- •Pineal Parenchymal Tumors
- •Germ Cell Tumors
- •Selected References
- •Medulloblastoma
- •Selected References
- •Anatomy of the Cranial Meninges
- •Meningomas
- •Primary Melanocytic Lesions
- •Other Related Neoplasms
- •Selected References
- •Cranial Nerve Anatomy
- •Schwannomas
- •Neurofibromas
- •Selected References
- •Histiocytic Tumors
- •Selected References
- •Sellar Region Anatomy
- •Normal Imaging Variants
- •Congenital Lesions
- •Neoplasms
- •Miscellaneous Lesions
- •Selected References
- •Intracranial Pseudotumors
- •Selected References
- •Metastatic Lesions
- •Paraneoplastic Syndromes
- •Selected References
- •Scalp Cysts
- •Extraaxial Cysts
- •Parenchymal Cysts
- •Intraventricular Cysts
- •Selected References
- •Anatomy and Physiology of the Basal Ganglia and Thalami
- •Selected References
- •Alcohol and Related Disorders
- •Opioids and Derivatives
- •Inhaled Gases and Toxins
- •Selected References
- •Selected References
- •Hypertensive Encephalopathies
- •Glucose Disorders
- •Thyroid Disorders
- •Seizures and Related Disorders
- •Miscellaneous Disorders
- •Selected References
- •The Normal Aging Brain
- •Dementias
- •Degenerative Disorders
- •Selected References
- •Normal Variants
- •Hydrocephalus
- •CSF Leaks and Sequelae
- •Selected References
- •Cerebral Hemisphere Formation
- •Imaging Approach to Brain Malformations
- •Posterior Fossa Anatomy
- •Chiari Malformations
- •Hindbrain Malformations
- •Selected References
- •Commissural Anomalies
- •Malformations Secondary to Abnormal Postmigrational Development
- •Selected References
- •Anencephaly
- •Holoprosencephaly
- •Holoprosencephaly Variants
- •Related Midline Disorders
- •Holoprosencephaly Mimics
- •Selected References
- •Selected References
- •Selected References
- •Cephaloceles
- •Craniosynostoses
- •Meningeal Anomalies
- •Selected References
- •Index

Toxic, Metabolic, Degenerative, and CSF Disorders
1036
(32-18) Embryonic thyroid migrates inferiorly from the tongue to the neck. Ectopic thyroid can occur anywhere along the thyroglossal duct .
thalamus is characteristic (32-17). In two-thirds of HIHH cases, T2* GRE or SWI demonstrates hypointense signal within the affected striatum. The signal abnormality probably represents manganese or zinc deposition, not calcium or hemorrhage.
Thyroid Disorders
Thyroid disorders are relatively common metabolic disturbances that are usually mild and rarely affect brain function. However, several imaging findings—some of them striking—have been associated with thyroid disease. Some can be mistaken for more serious disease (e.g., hypothyroidinduced pituitary hyperplasia mimicking pituitary adenoma), and a few (e.g., Hashimoto encephalopathy) can be life-threatening.
Hypothyroidism can be congenital or acquired. We begin our discussion of thyroid disease with congenital hypothyroidism before turning our attention to acquired hypothyroid disease and its imaging manifestations. We close the section with a brief consideration of hyperthyroid disease and its effects on the CNS.
(32-19) Graphic depicts possible sites of ectopic thyroid along the thyroglossal duct, but they may also occur elsewhere (e.g., substernal).
Congenital Hypothyroidism
Hypothyroidism is one of the most frequent congenital endocrine disorders. Congenital hypothyroidism (CH) occurs in 1:2,000-4,000 newborns and is one of the most common preventable causes of mental retardation. If the diagnosis is made and treatment begun within a few weeks of birth, neurodevelopmental outcome is generally normal.
Newborns with CH normally have some initial residual thyroid function because maternal T4 crosses the placenta to the fetus. With a half-life of 6 days, however, maternal T4 will be almost completely metabolized and excreted by 3-4 weeks of age.
In developed countries with newborn screening programs, most infants with CH are diagnosed soon after birth. Serum TSH is elevated (typically more than 20-30 mU/L). In less developed countries, CH is often diagnosed later in childhood when suspicious clinical features lead to serum thyroid function testing or imaging studies. The most visible clinical feature is a facies suggestive of "cretinism," a term no longer used because of its pejorative implications.
(32-20) Graphic depicts hypothalamic-pituitary- thyroid axis. T3 and T4 inhibit hypothalamic and pituitary stimulation of thyroid.
Etiology and Presentation
CH can be caused by thyroid dysgenesis, dyshormonogenesis, or central hypothyroidism. Iodine deficiency and maternal thyroid disease can also cause hypothyroidism in neonates.
Maternal Factors. Transient CH can occur in preterm infants born in areas with endemic iodine deficiency or to families with a history of goiter.
In maternal autoimmune thyroiditis, IgG antibodies cross the placenta and may block fetal thyroid production. Medication or I-131 therapy for maternal hyperthyroidism or cancer can also act adversely on the fetus.
Thyroid Dysgenesis. Thyroid dysgenesis is the most common cause of CH, accounting for 70-75% of cases. Failure of normal thyroid gland development includes both abnormal gland formation and aberrant thyroid descent.
Ectopic thyroid tissue accounts for 25-50% of cases with thyroid dysgenesis. Ectopia can occur anywhere along the embryonic thyroglossal duct (32-18), the path that the developing thyroid follows as it descends from the tongue base to the neck (32-19). Hormone production in ectopic thyroids is low
Acquired Metabolic and Systemic Disorders
(despite the presence of functioning tissue) but not completely absent. In some cases, hormone production may be enough to delay clinical symptoms until adolescence.
Thyroid agenesis or hypoplasia accounts for 20-50% of CH cases and typically causes severe hypothyroidism with markedly depressed T4, elevated TSH, and undetectable levels of thyroglobulin.
Dyshormonogenesis. Inborn errors of thyroid hormone biosynthesis (dyshormonogenesis) account for 5-15% of CH cases. Here, defects occur in the biosynthesis, secretion, or utilization of thyroid hormone. These include enzymatic abnormalities, deficient iodide trapping due to sodium-iodide symporter defects, TSH resistance with abnormal TSH receptors on the follicular cell membranes, and peripheral thyroid hormone resistance with T3 receptor defects in peripheral cell nuclei.
Central (Secondary) Hypothyroidism. Pituitary or hypothalamic dysfunction causes 10-15% of CH cases. The gland is formed normally, but the hypothalamic-pituitary-thyroid axis is disrupted (32-20). Most cases of secondary hypothyroidism are caused by decreased pituitary TSH and often occur in the setting of combined pituitary hormone deficiency. So-called tertiary hypothyroidism from low hypothalamic TSH is rare in children.
Imaging
Imaging studies of the face and neck demonstrate an ectopic thyroid in up to half of all CH cases. Nearly 90% occur at the tongue base, where they are seen as a well-delineated, round or ovoid, hyperdense midline mass on NECT and a T1 iso-/T2 hyperintense mass on MR. Avid uniform enhancement following contrast administration is typical.
Complete absence of the thyroid and thyroid hypoplasia accounts for most of the remaining cases. Although high-resolution ultrasound is considered the "gold standard" for measuring thyroid dimensions, it lacks sensitivity for detecting small ectopic glands and is often combined with scintigraphy—considered the "gold standard"—for the work-up of CH.
Nuclear medicine studies (Tc-99m pertechnetate or I-123 scans) are typically used to diagnose thyroid agenesis and demonstrate absent uptake (no activity in any of the expected sites) (32-21). Inborn errors of thyroid hormone metabolism often appear as a small bilobed thyroid in the expected location (32-22).
Brain MR in children with CH may show mild generalized atrophy with reduced WM volume and poor differentiation of cortical layers, enlarged sylvian fissures, and T2 hypointensity in the globi pallidi and substantia nigra.
Differential Diagnosis
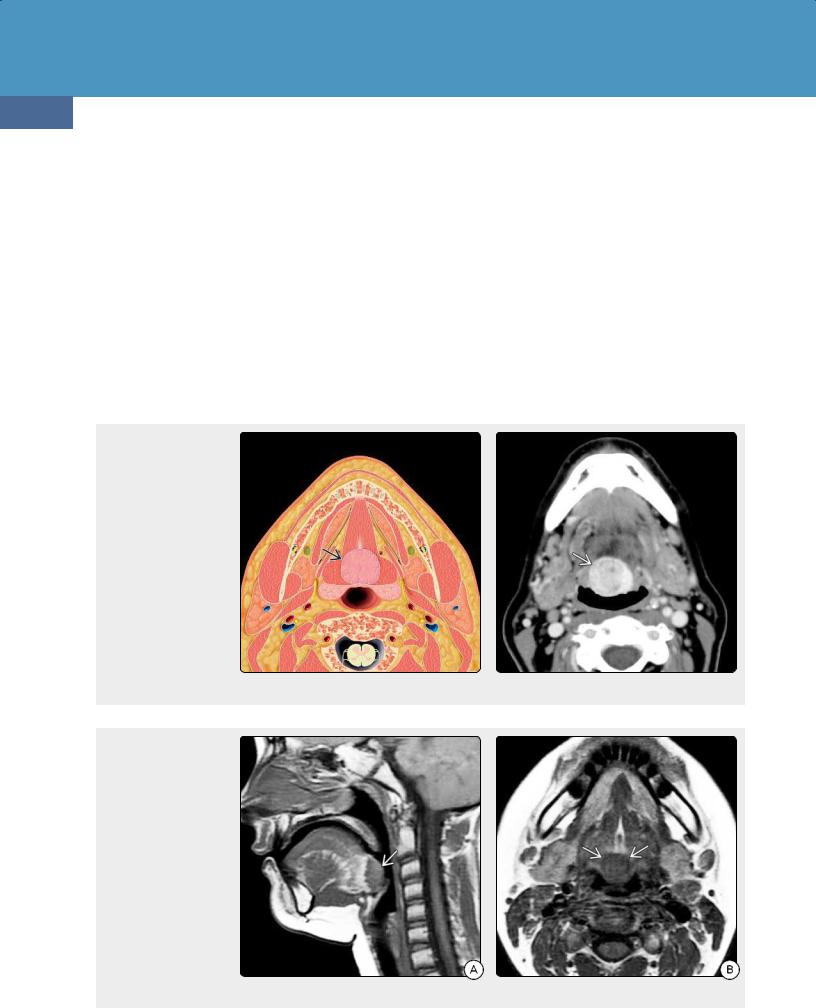
As the central tongue base is the most common location for an ectopic thyroid (32-24), the most important differential diagnoses are venous malformation or hemangioma, prominent/asymmetric lingual tonsil, and neoplasm (non-Hodgkin lymphoma). A lingual thyroid may expand dramatically during puberty. In 75% of cases, a lingual thyroid is the only functioning thyroid tissue; it must not be mistaken for tumor and removed (3225) (32-26)!
Venous malformation exhibits prominent T2 hyperintensity. An upper airway infantile hemangioma is usually subglottic and asymmetric. A transglottic hemangioma involves multiple structures, not just the tongue base. Prominent/asymmetric tonsillar tissue has the same density/signal intensity as other lymphoid structures.
1037
(32-21) Tc-99m in congenital hypothyroidism shows no uptake in oropharynx, tongue base , neck , mediastinum . (J. P. O'Malley, MD.)
(32-22) Hypothyroidism secondary to organification defect with bilobed thyroid shows very low uptake . (J. P. O'Malley, MD.)
(32-23) (L) T1 C+ shows 8y boy with hyperplasiainitially called pituitary macroadenoma. (R) After PTH, pituitary is normal.
Toxic, Metabolic, Degenerative, and CSF Disorders
1038
Acquired Hypothyroid Disorders
Acquired hypothyroidism is much more common than the congenital variety, affecting eight to nine million Americans and many more patients worldwide. Acquired hypothyroidism has two important imaging manifestations: pituitary hyperplasia and Hashimoto thyroiditis/encephalopathy.
Pituitary Hyperplasia
Enlarged pituitary glands are common in young menstruating female patients and pregnant/lactating female patients. Nonphysiologic increase in pituitary volume—pathologic pituitary enlargement—is much less common and typically occurs in response to end-organ failure.
Hypothyroidism can result in secondary TSH cell hyperplasia, symmetrically enlarging the pituitary gland and mimicking a pituitary adenoma on imaging studies. Both physiologic and nonphysiologic pituitary hyperplasia are discussed in detail in
(32-24) Axial graphic depicts lingual thyroid in the posterior midline of the tongue, deep to the foramen cecum. Sharply defined contour and midline location at the tongue base or floor of the mouth are typical of lingual thyroid. (32-25) CECT scan shows classic lingual thyroid , seen as a uniformly enhancing midline mass in the base of the tongue.
(32-26A) Sagittal T1WI in a 13y hypothyroid girl shows a well-delineated mass at the tongue base. The mass is isointense with the intrinsic musculature of the tongue. (32-26B) Axial T1WI in the same patient shows that the mass is midline and sharply demarcated. This is classic lingual thyroid.
Chapter 25. Most cases of hypothyroid-induced pituitary hyperplasia reverse with thyroid hormone replacement therapy (32-23). Caution: Any prepubescent male patient thought to harbor a "pituitary macroadenoma" on imaging studies should undergo comprehensive endocrine evaluation, as macroadenomas are exceptionally rare in this age group!
Hashimoto Encephalopathy
Terminology and Etiology. Hashimoto encephalopathy is a rare but treatable condition typically associated with Hashimoto thyroiditis and characterized by high levels of antithyroid antibodies. Hashimoto encephalitis is also called "steroid-responsive encephalopathy with autoimmune thyroiditis." It is a well-recognized neurologic complication of autoimmune thyroid disease and is the most common cause of acquired hypothyroidism.
Occasionally, Hashimoto encephalopathy occurs with severe "iatrogenic" hypothyroidism, typically with inadequate
Acquired Metabolic and Systemic Disorders
1039
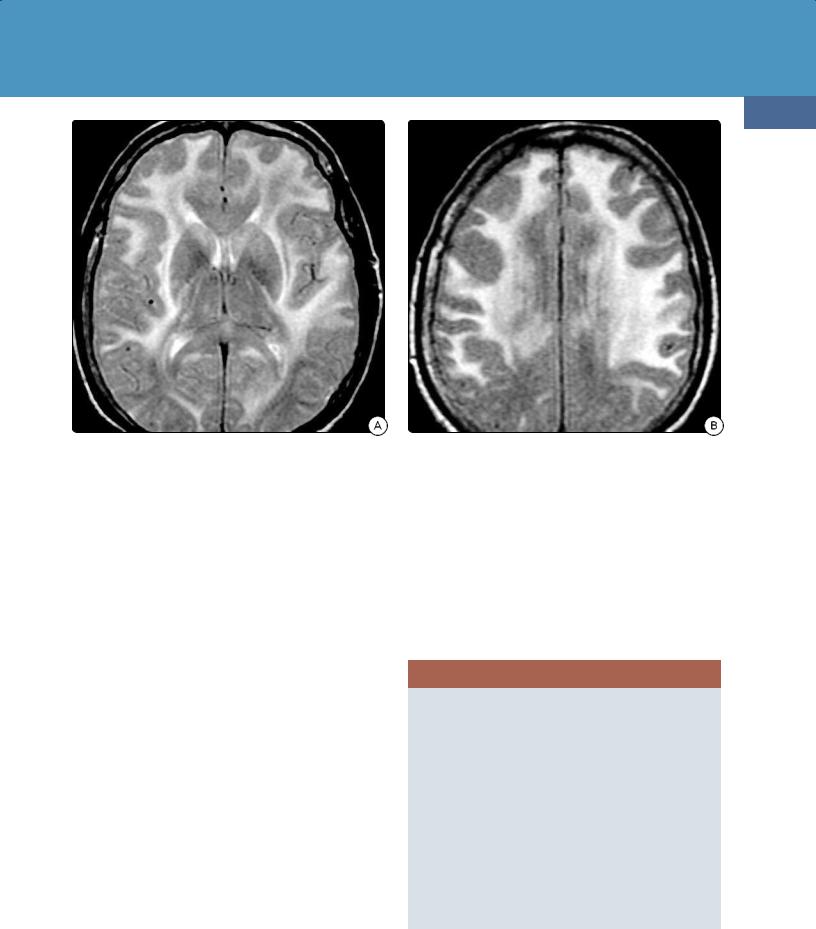
(32-27A) Axial T2WI in a 58y woman with acute Hashimoto encephalopathy shows confluent, symmetric hyperintensity in the subcortical, deep WM.
(32-27B) FLAIR scan in the same patient shows involvement of the frontal subcortical WM with marked sparing of the occipital lobes.
hormone replacement following thyroidectomy or radioactive I-131 treatment.
Clinical Issues. Hashimoto encephalopathy occurs in both children and adults. There is a moderate female predominance.
Most patients present with acute encephalopathy or severe neuropsychiatric disturbances (sometimes termed "myxedema madness"). Other common symptoms include seizures (66%), myoclonus (38%), and stroke (27%). Occasionally, patients present with more gradual cognitive decline and personality changes. These patients may be initially misdiagnosed as having "presenile" dementia.
Imaging. Approximately half of all patients with Hashimoto encephalopathy demonstrate imaging abnormalities. The most typical MR findings are diffuse confluent or focal T2/FLAIR hyperintensities in the subcortical and deep periventricular white matter. The occipital lobes are relatively spared (32-27). Hashimoto encephalopathy typically does not enhance following contrast administration.
Hyperthyroidism
The most common manifestation of hyperthyroidism in the head and neck is thyroid ophthalmopathy (Graves disease). Brain involvement in hyperthyroidism occurs but is very rare.
Thyrotropin-secreting pituitary macroadenomas cause hyperthyroidism due to the syndrome of inappropriate TSH secretion. Hypocortisolemic states (e.g., Sheehan syndrome with perior postpartum pituitary necrosis) can also precipitate hyperimmunity and autoimmune hyperthyroidism.
There is an increased prevalence of psychiatric and behavioral disturbances in patients with hyperthyroidism, including apathetic hyperthyroidism and hyperthyroid dementia. Thyrotoxicosis can also cause disturbances of consciousness. Seizures, usually the generalized tonic-clonic type, occur in less than 1% of cases.
Brain imaging in hyperthyroidism is uncommon. A few cases of acute idiopathic intracranial hypertension ("pseudotumor cerebri") associated with hyperthyroidism have been reported.
THYROID DISORDERS
Congenital Hypothyroid Disorders
•Etiology
○Thyroid dysgenesis, dyshormonogenesis, central hypothyroidism
•Imaging
○Ectopic thyroid in 50% (90% at base of tongue)
○Do not mistake for neoplasm!
Acquired Hypothyroid Disorders
•Pituitary hyperplasia
○Prepubescent male patient with "fat" pituitary
•Hashimoto encephalopathy
○WM edema (spares occipital lobes)
Hyperthyroidism
•Thyroid ophthalmopathy (Graves disease)
•Brain rarely imaged
Because of its effect on factor VIII activity, hyperthyroidism has also been reported as an independent risk factor for dural venous sinus thrombosis. Graves disease has been reported as a rare cause of transient corpus callosum splenium
Toxic, Metabolic, Degenerative, and CSF Disorders
1040
(32-28) NECT scan of the skull in a patient with HPTH shows the characteristic alternating "salt and pepper" foci of resorption and sclerosis.
(32-29A) NECT in a 54y man with hyperparathyroidism shows extensive symmetric Ca++ in basal ganglia , thalami , cortex .
(32-29B) Coronal NECT in the same case shows how symmetric the basal Ca++ is. Note GM-WM interface calcifications .
hyperintensity and an MS-like multiphasic demyelinating autoimmune syndrome.
Parathyroid and Related
Disorders
The parathyroid glands lie in the visceral space of the neck. They are normally the size and shape of kidney beans and are closely adherent to the posterior surfaces of the thyroid lobes. Ectopic parathyroid glands are found in 2% of cases, typically just below the inferior thyroid pole, although they can be found from the upper neck to the mediastinum. Most patients have four parathyroid glands, 10% have five or more, and 3% have three or fewer glands.
Metabolic abnormalities related to parathyroid hormone dysfunction include primary and secondary hyperparathyroidism as well as hypoparathyroidism, pseudohypoparathyroidism, and pseudo-pseudohypoparathyroidism.
Hyperparathyroidism
The parathyroid glands control calcium metabolism by producing parathyroid hormone (PTH). Hyperparathyroidism (HPTH) is the classic disease of bone resorption, so imaging abnormalities may be seen in both the skull and brain.
HPTH can be an acquired (common) or inherited disorder (rare). Both conditions are briefly discussed in this chapter. HPTH can also be primary, secondary, or even tertiary. Because of the increasing number of patients on dialysis, the most common type is now secondary HPTH.
Primary Hyperparathyroidism
Etiology. In primary HPTH (1° HPTH), excessive levels of PTH result in unneeded bone resorption. The most common cause of 1° HPTH is parathyroid adenoma, responsible for 75-85% of cases. The second most common etiology is nonneoplastic parathyroid hyperplasia (10-20%). Parathyroid carcinoma is rare, accounting for just 1-5% of cases.
Sporadic 1° HPTH is much more common than hereditary HPTH. The most important inherited syndromes associated with 1° HPTH are multiple endocrine neoplasia (MEN) type 1, MEN type 2A, and familial isolated HPTH. Major features in patients with MEN1 include parathyroid tumor (95%), pancreatic neuroendocrine tumor (40%), and pituitary neoplasm (30%). MEN2A is characterized by medullary thyroid carcinoma (99%), pheochromocytoma (50%), and parathyroid tumors (20-30%).
Clinical Issues. 1° HPTH is most common in middle-aged to older adults and relatively rare in children. There is a striking female predominance. 1° HPTH is characterized by hypercalcemia and hypophosphatemia (serum calcium is elevated; serum phosphorus is normal or decreased). HPTH is usually asymptomatic. General signs of symptomatic HPTH have been characterized as "stones, bones, abdominal groans, and psychic moans."
A recently described (and controversial) entity is called "normocalcemic primary hyperparathyroidism." Patients have elevated PTH levels but normal serum calcium and vitamin D levels.
Imaging. Standard two-phase contrast-enhanced CT has an overall pooled sensitivity of 73% and PPV of 81% for localization of the pathologic parathyroid gland to the correct quadrant. 4D-CT performs comparably well to US and sestamibi scans, with an overall sensitivity of 80-90%.

Acquired Metabolic and Systemic Disorders
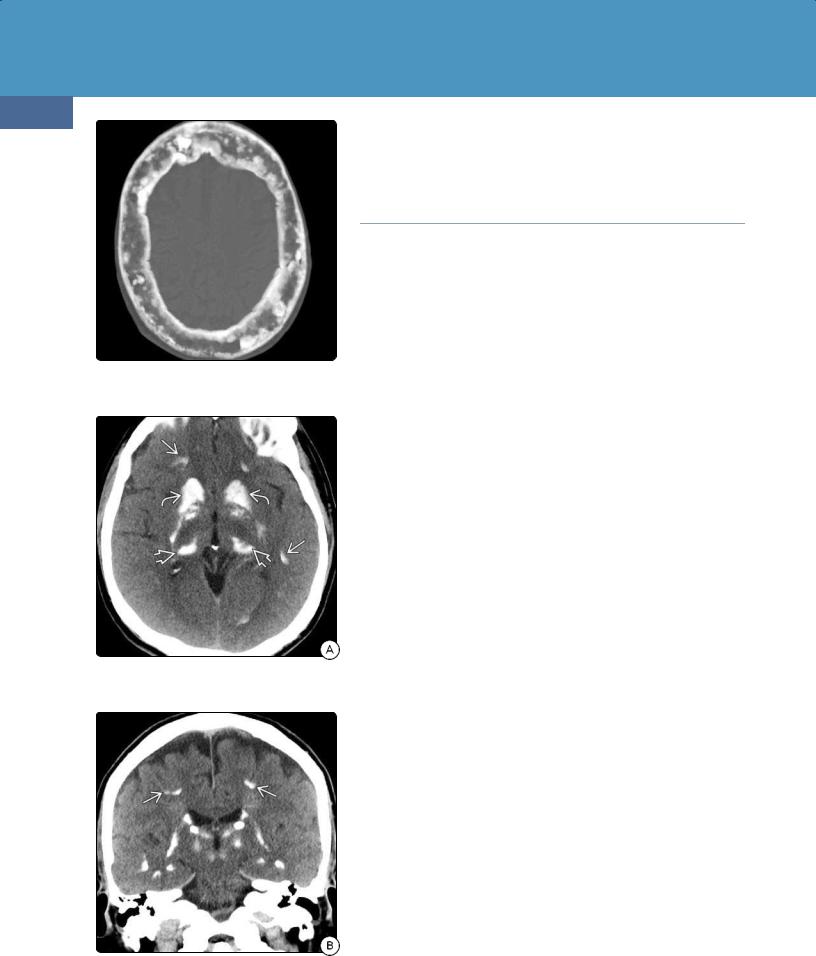
Bone CT demonstrates diffuse patchy "salt and pepper" lesions in the skull. These are caused by foci of bone resorption interspersed with variable patchy sclerosis (32-28). Bilateral symmetric resorption of the lamina dura of the teeth may be present.
The most common findings in the brain are basal ganglia calcifications on NECT. Bilateral symmetric deposits in the globi pallidi, putamen, and caudate nuclei are typical. The thalami, subcortical WM, and dentate nuclei may also be affected (32-29).
MR shows symmetric T1 shortening and T2 hypointensity in the basal ganglia. Mild to moderate "blooming" on T2* (GRE, SWI) sequences is typical. "Brown tumors"—solitary or multiple nonneoplastic lesions in the skull—are common (see below).
Secondary Hyperparathyroidism
Etiology. Secondary HPTH (2° HPTH) is characterized by PTH hypersecretion and parathyroid gland hyperplasia.
The most common cause of 2° HPTH is chronic renal disease (CRD). The majority of dialysis patients eventually develop 2° HPTH. Other etiologies of 2° HPTH include dietary calcium deficiency, vitamin D disorders, disrupted phosphate metabolism, and hypomagnesemia.
Clinical Issues. Most patients with 2° HPTH are older than 40 years at the time of initial diagnosis. There is no sex predilection. Serum calcium is normal or low, serum phosphorus is increased, and calcium-phosphate product is elevated. Vitamin D is low, almost always secondary to renal disease rather than dietary deficiency.
A common manifestation of CRD is renal osteodystrophy. Massive thickening of the calvaria and skull base narrows neural and vascular channels. Progressive cranial nerve involvement—most commonly compressive optic neuropathy—and carotid stenosis with ischemic symptoms are typical.
Imaging. 2° HPTH primarily affects the skull and dura; the brain parenchyma itself is usually normal. NECT scans show markedly thickened skull and facial bones, a condition sometimes referred to as "uremic leontiasis ossea" or "big head disease" (32-30).
"Brown tumors" can be seen in both 1° HPTH and 2° HPTH. "Brown tumors" represent a reactive—not neoplastic—process caused by osteoclastic bone resorption. Fibrous replacement, hemorrhage, and necrosis lead to formation of brownish-appearing cysts. Solitary or multiple "brown tumors" are seen on bone CT as focal expansile lytic lesions with nonsclerotic margins. Signal intensity on MR is highly variable, reflecting the age and amount of hemorrhage as well as the presence of fibrous tissue and cyst formation (32-32).
The classic intracranial finding in 2° HPTH is unusually extensive, plaque-like dural thickening (32-31). Longstanding CRD can also result in extensive "pipestem" calcifications in the internal and external carotid arteries.
Tertiary Hyperparathyroidism
Tertiary HPTH (3° HPTH) results from longstanding 2° HPTH. Full-blown tertiary HPTH is rarely seen. The parathyroid gland becomes hyperplastic and does not respond appropriately to serum calcium levels (i.e., functions "autonomously"). Imaging findings are similar to those of 2° HPTH.
1041
(32-30A) Axial bone CT in a patient with 2° HPTH shows leontiasis ossea with marked calvarial thickening, focal sclerotic "brown tumors" .
(32-30B) Coronal bone CT in the same patient demonstrates the striking calvarial thickening.
(32-31) NECT scan in a 31y man with ESRD shows markedly thickened, plaque-like deposits along the tentorium .
Toxic, Metabolic, Degenerative, and CSF Disorders
1042
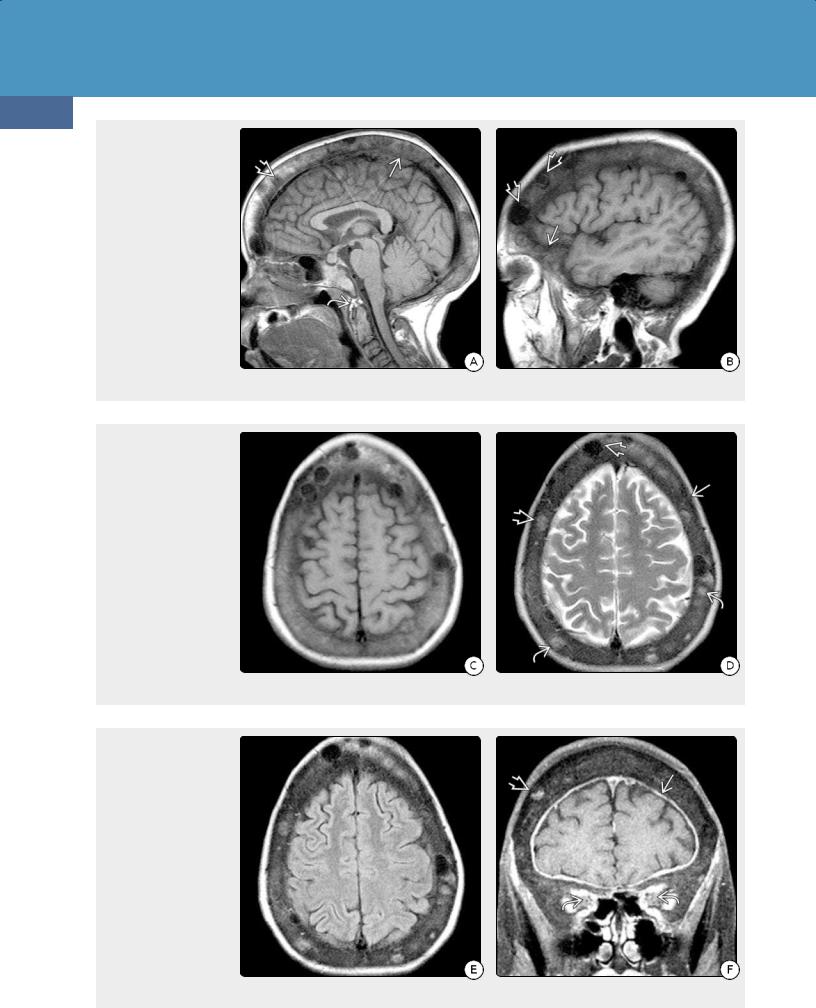
(32-32A) Sagittal T1WI shows a 34y man with 25- y history of renal failure on dialysis, known 2° HPTH, and "big head disease." He developed decreasing vision, swollen optic discs. Note markedly thickened skull with multiple "brown tumors", thickened calcified pannus around the odontoid . (32-32B) Sagittal T1WI in same patient shows thickening of orbital wall and calvaria. Note welldelineated "brown tumors" of varying SI .
(32-32C) Axial T1WI shows the markedly thickened calvaria and focal lesions. (32-32D) T2WI shows thick calvaria, multiple "brown tumors" of varying signal intensity . Note that some of the more hyperintense lesions are virtually invisible on the T1WI to the left.
(32-32E) FLAIR scan shows the thick skull, multifocal "brown tumors." The underlying brain appears normal. (3232F) Coronal T1 C+ FS shows that one of the "brown tumors" enhances. Note thick dura . The thickened bone causes significantly decreased volume of the orbits with compression of the optic nerve sheaths at the orbital apex. (All six images courtesy S. Chung, MD.)

Acquired Metabolic and Systemic Disorders
Hypoparathyroid Disorders
Three types of hypoparathyroidism are recognized: hypoparathyroidism, pseudohypoparathyroidism, and pseudopseudohypoparathyroidism. All three disorders share common features on brain imaging although their clinical presentation and laboratory findings vary.
Hypoparathyroidism
Childhood hypoparathyroidism (HP) usually presents around age five. It is probably an autoimmune-mediated disease with decreased parathyroid hormone (PTH) production. HP is characterized by hypocalcemia and hyperphosphatemia. Carpal-pedal spasm, tetany, seizures, and hyperreflexia are common presentations.
Ectopic calcification in brain tissue is one of the characteristic typical findings in HP on NECT scans (32-33). The basal ganglia and thalami are the most common sites, followed by the
1043
cerebrum and cerebellum. Subcutaneous soft tissue calcifications are common in the extremities but rare in the head and neck.
The most striking extracranial imaging findings are related to osteosclerosis. Spinal ligament calcification/ossification, osteophyte formation, and enthesopathy (especially around the pelvis) are typical.
Adult HP is rare. The most common causes in adults are parathyroid gland injury and inadvertent removal during neck dissection in thyroid cancer, which carries a 37% risk of permanent HP.
Complications of chronic hypocalcemia in adults include renal dysfunction, nephrocalcinosis, kidney stones, basal ganglia calcifications, and posterior subcapsular cataracts, as well as low bone turnover and increased bone density.
(32-33A) Axial NECT scan in a 7y patient with new onset of seizures and documented hypoparathyroidism shows symmetric calcifications in the globi pallidi with smaller calcific foci at the GM-WM interfaces . (32-33B) Slightly more cephalad scan shows additional calcifications.
(32-34A) Axial T1WI in a
34y woman with PPHP on calcitriol shows symmetric T1 shortening in both caudate nuclei and putamina . (32-34B) T2* SWI in the same case shows symmetric hypointensity in both caudate nuclei , putamina , and the globi pallidi . (Courtesy P. Hildenbrand, MD.)

Toxic, Metabolic, Degenerative, and CSF Disorders
1044
Pseudohypoparathyroidism and PseudoPseudohypoparathyroidism
Both pseudohypoparathyroidism (PHP) and pseudopseudohypoparathyroidism (PPHP) are caused by mutations in GNAS and STX16, which confer resistance to PTH. In both disorders, the parathyroid glands produce PTH.
PHP is characterized by elevated PTH levels and PTH-resistant hypocalcemia and hyperphosphatemia. Obesity, round face, and mental retardation are the key clinical features. Albright hereditary osteodystrophy is a specific phenotype seen in autosomal-dominant PHP and is characterized by short fourth and fifth metacarpals and short stature.
PPHP is characterized by incomplete expression of PHP (hence the term "pseudo-pseudo"). PPHP typically shows no laboratory abnormalities, so calcium and phosphate levels are normal.
Bilateral symmetric calcifications in the basal ganglia and thalami, cerebellar hemispheres, subcortical WM, and occasionally the cerebral cortex are typical findings in both PHP and PPHP (32-34).
PARATHYROID DISORDERS
Hyperparathyroidism
•1° hyperparathyroidism (parathyroid adenomas)
○"Salt and pepper" skull, "brown tumors"
○Basal ganglia Ca++
•2° (chronic renal failure)
○Thick skull, face ("big head" disease) ± brown tumors
○Plaque-like dural thickening, Ca++
Hypoparathyroid Disorders
•3 types (distinguished by clinical, laboratory findings)
○Hypoparathyroidism
○Pseudohypoparathyroidism
○Pseudo-pseudohypoparathyroidism
•All have Ca++ in basal ganglia > cerebrum, cerebellum
Primary Familial Brain Calcification
(Fahr Disease)
Primary familial brain calcification (PFBC) is an inherited disorder that results in striking brain calcifications. Once thought to be idiopathic, it is included in this chapter (rather than Chapter 31 on inherited metabolic disorders), as its imaging findings closely resemble those seen in pseudohypoparathyroidism and pseudopseudohypoparathyroidism.
Terminology
PFBC, formerly termed Fahr disease, has also been termed idiopathic basal ganglia calcification and bilateral striopallidodentate calcinosis. PFBC is characterized by basal ganglia and extraganglionic calcifications, parkinsonism, and neuropsychiatric symptoms.
Etiology
PFBC is caused by mutations in four genes, namely SLC20A2,
PDGFB, PDGFRB, and XPR1.
Pathology
Autopsies of the few described cases of PFBC disclose severe brain calcification in the basal ganglia, thalami, cerebral WM, cerebellar WM, and dentate nuclei. Severe cases show rows of small calcospherites along capillaries. In some cases, diffuse neurofibrillary tangles with Fahr-type calcification have been identified in patients with early-onset Alzheimer dementia.
Clinical Issues
Fahr disease is typically asymptomatic in the first and second decades. There is no sex predilection. Calcium-phosphorus metabolism and PTH levels are normal.
Deposition of calcium, along with other minerals, typically begins in the third decade, but symptoms develop one or two decades later, usually between ages 30 and 60 years. Clinical findings follow a bimodal distribution; schizophrenic-like psychosis typically presents in early adulthood with extrapyramidal symptoms, and subcortical dementia predominates in patients over the age of 50.
Imaging
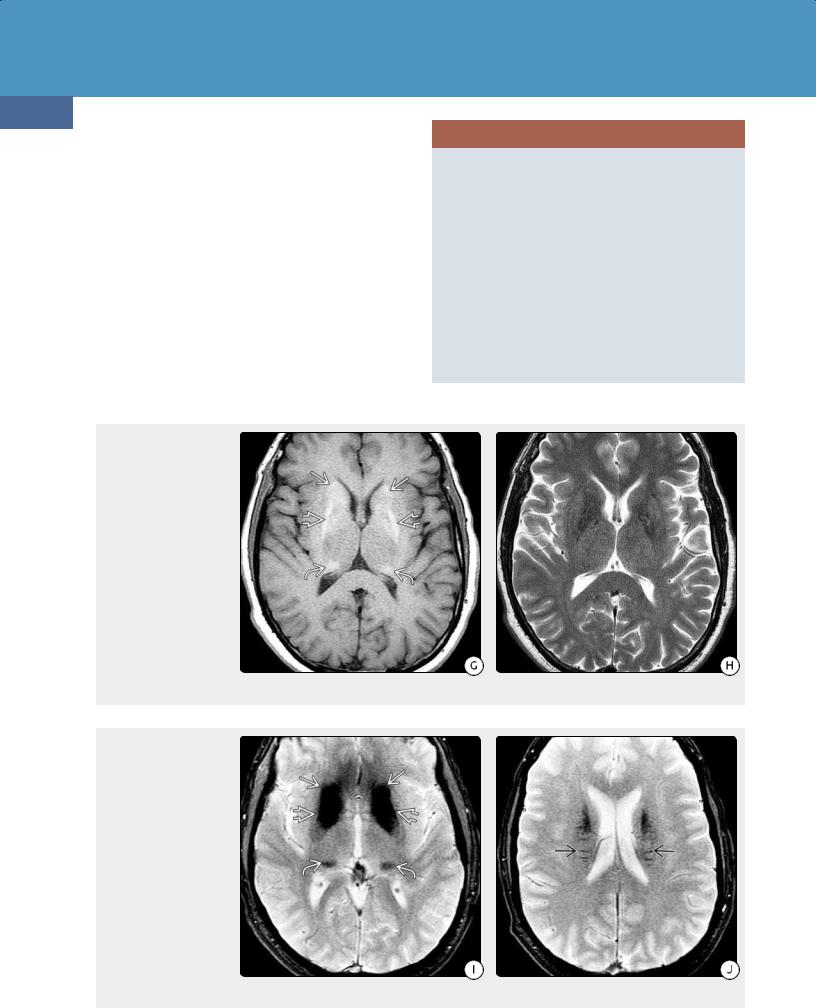
CT Findings. NECT discloses extensive bilateral, relatively symmetric basal ganglia calcification in virtually all cases of PFBC. The lateral globus pallidus (GP) is the most severely affected with relative sparing of the medial GP. The putamen, caudate, thalami, dentate nuclei of the cerebellum, and both the cerebral and cerebellar WM (including the internal capsule) are commonly affected (32-35). Thalamic and dentate nucleus calcification is most frequently associated with SLC20A2 mutation.
PFBC does not enhance on CECT.
MR Findings. MRs in PFBC can be confusing. Signal intensity varies according to disease stage and the amount of calcification and heavy metal deposition. Calcification is typically hyperintense on T1WI but can be quite variable on T2WI (32-35G) (32-35H).
T2/FLAIR scans may appear normal or mildly abnormal. They may also show extensive foci of T2 prolongation in the cerebral WM that can be so striking as to mimic toxic/metabolic demyelination (32-36). The pattern of WM cysts with leukodystrophy seems to be a marker for PDGFB mutation.
T2* (GRE, SWI) scans show profound susceptibility changes with "blooming" hypointensity secondary to iron deposition (32-35I) (32-35J). Fahr disease does not enhance on T1 C+ sequences.
Ultrasound. Transcranial sonography performed through the temporal bone acoustic windows demonstrates increased echogenicity in the basal ganglia, thalami, and substantia nigra.

Acquired Metabolic and Systemic Disorders
1045
(32-35A) Series of axial NECT scans in a 51y man with Fahr disease shows bilaterally symmetric calcifications in the cerebellar white matter. (32-35B) NECT scan shows very dense calcifications in both caudate nuclei and globi pallidi , as well as more faint calcification in the frontal white matter .
(32-35C) More cephalad NECT scan in the same patient shows calcification in the putamina and lateral globi pallidi , with relative sparing of the most medial GP . Calcification is present in the pulvinars of both thalami . Punctate calcification is seen in the cerebral WM. (32-35D) More extensive calcification is present in the caudate heads and bodies.
(32-35E) NECT scan shows linear calcification extending perpendicularly from the caudate nuclei into the cerebral white matter . (32-35F) NECT scan through the corona radiata shows innumerable faint linear calcifications extending throughout the deep cerebral WM .
Toxic, Metabolic, Degenerative, and CSF Disorders
1046
Nuclear Medicine. SPECT scans have demonstrated increased uptake in the temporal lobes with decreased basal ganglia uptake. This may reflect hyperactivation with disruption of the cortical-subcortical neural circuits responsible for the psychotic episodes often associated with Fahr disease.
Differential Diagnosis
Basal ganglia calcification is nonspecific and can be physiologic or the end result of a variety of toxic, metabolic, inflammatory, and infectious insults. Specific sublocation of the calcification can be very helpful in determining its underlying etiology.
The major differential diagnosis of PFBC is normal physiologic calcification of the basal ganglia. Age-related ("senescent") calcification in the basal ganglia is common, typically localized in the medial GP, relatively minor, and of no clinical significance. PFBC has much heavier, far more extensive calcification.
PRIMARY FAMILIAL BRAIN CALCIFICATION
Pathoetiology, Clinical Features
•Also known as Fahr disease
•Caused by 4 gene mutations (SLC20A2 most common)
•Usually presents between 30 and 60 years
○Extrapyramidal symptoms, dementia
Imaging Findings
•NECT
○Extensive bilateral BG Ca++
○Putamen, caudate, thalami, dentate nuclei
○WM of hemispheres, cerebellum
•MR
○T1 shortening in areas of calcification
○± T2/FLAIR WM hyperintensity, cysts
○Extensive "blooming" on T2* (GRE/SWI)
○DDx = physiological Ca++, PHP/PPHP
(32-35G) Axial T1WI in the same patient as shown on the previous page shows relatively symmetric hyperintensity in the caudate heads , lateral globi pallidi , and pulvinars of both thalami. (32-35H) T2WI in the same patient shows no visible abnormalities.
(32-35I) T2* GRE scan shows striking "blooming" in the same areas (caudate heads , globi pallidi , and thalamic pulvinars ) as the calcifications on NECT scans and corresponding hyperintensities on the T1WI above. (32-35J) T2* GRE scan shows linear "blooming" foci extending perpendicularly from the caudate nuclei into the deep periventricular white matter .