

- •Pineal Parenchymal Tumors
- •Germ Cell Tumors
- •Selected References
- •Medulloblastoma
- •Selected References
- •Anatomy of the Cranial Meninges
- •Meningomas
- •Primary Melanocytic Lesions
- •Other Related Neoplasms
- •Selected References
- •Cranial Nerve Anatomy
- •Schwannomas
- •Neurofibromas
- •Selected References
- •Histiocytic Tumors
- •Selected References
- •Sellar Region Anatomy
- •Normal Imaging Variants
- •Congenital Lesions
- •Neoplasms
- •Miscellaneous Lesions
- •Selected References
- •Intracranial Pseudotumors
- •Selected References
- •Metastatic Lesions
- •Paraneoplastic Syndromes
- •Selected References
- •Scalp Cysts
- •Extraaxial Cysts
- •Parenchymal Cysts
- •Intraventricular Cysts
- •Selected References
- •Anatomy and Physiology of the Basal Ganglia and Thalami
- •Selected References
- •Alcohol and Related Disorders
- •Opioids and Derivatives
- •Inhaled Gases and Toxins
- •Selected References
- •Selected References
- •Hypertensive Encephalopathies
- •Glucose Disorders
- •Thyroid Disorders
- •Seizures and Related Disorders
- •Miscellaneous Disorders
- •Selected References
- •The Normal Aging Brain
- •Dementias
- •Degenerative Disorders
- •Selected References
- •Normal Variants
- •Hydrocephalus
- •CSF Leaks and Sequelae
- •Selected References
- •Cerebral Hemisphere Formation
- •Imaging Approach to Brain Malformations
- •Posterior Fossa Anatomy
- •Chiari Malformations
- •Hindbrain Malformations
- •Selected References
- •Commissural Anomalies
- •Malformations Secondary to Abnormal Postmigrational Development
- •Selected References
- •Anencephaly
- •Holoprosencephaly
- •Holoprosencephaly Variants
- •Related Midline Disorders
- •Holoprosencephaly Mimics
- •Selected References
- •Selected References
- •Selected References
- •Cephaloceles
- •Craniosynostoses
- •Meningeal Anomalies
- •Selected References
- •Index
Holoprosencephalies, Related Disorders, and Mimics
1237
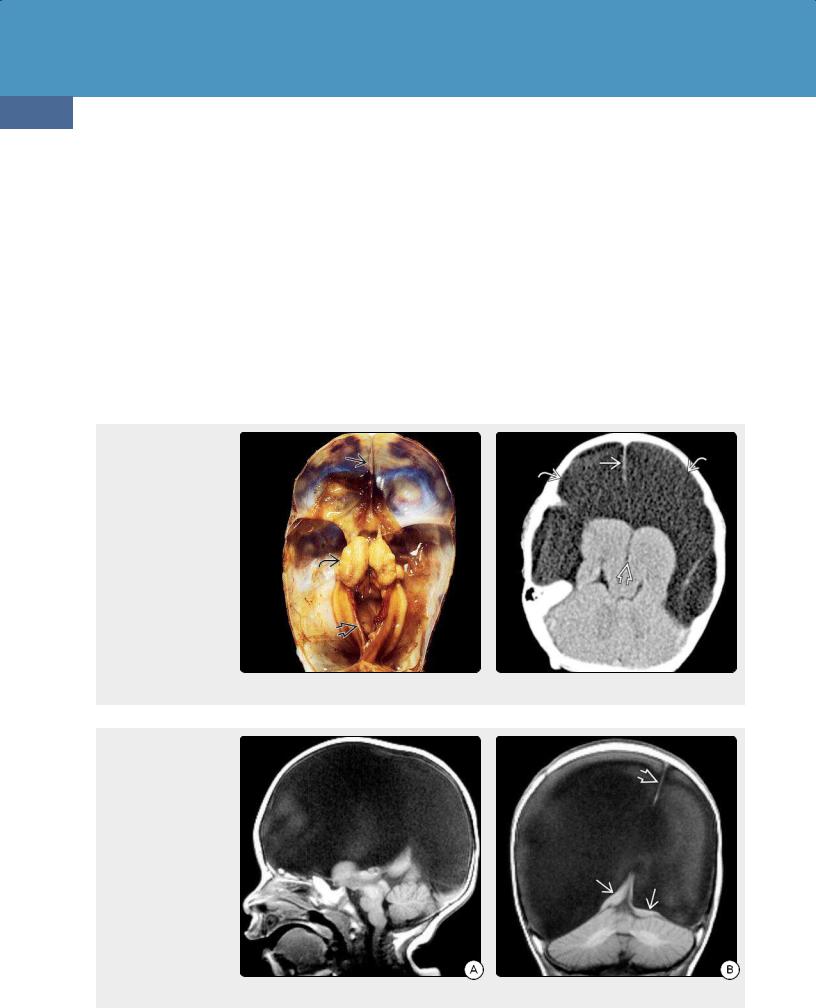
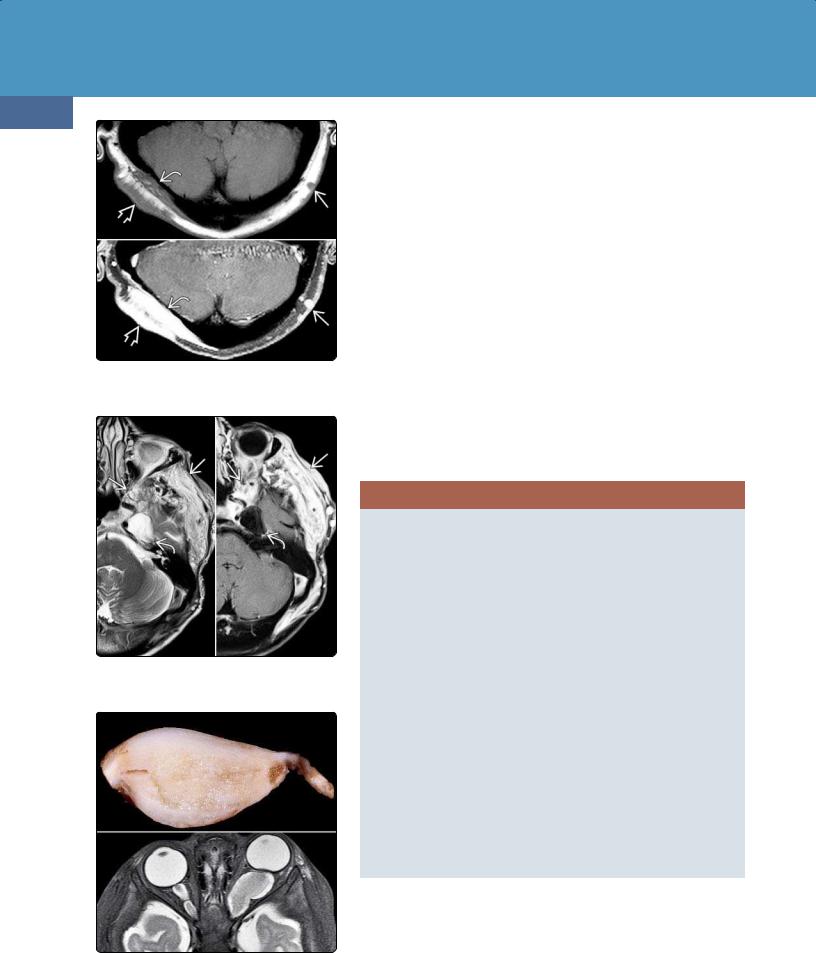
(38-22A) Autopsy case of hydranencephaly demonstrates a large head with striking transillumination indicating that most of the cranium is water-filled.
choanal atresia, developmental retardation, genital anomalies, ear anomalies) syndrome.
Holoprosencephaly Mimics
Hydranencephaly
Although some authors consider hydranencephaly a congenital malformation, it is actually the consequence of severe brain destruction in utero. We discuss it here, as it is important to recognize and distinguish hydranencephaly from other disorders such as alobar holoprosencephaly or maximal hydrocephalus.
Terminology
The term hydranencephaly is a contraction of "hydroanencéphalie" and literally means water without the brain. In hydranencephaly, the cerebral hemispheres are completely or almost completely missing. Instead, a membranous sac filled with CSF, glial tissue, and ependyma is present. In rare instances, only one hemisphere is destroyed. This condition is termed hemihydranencephaly.
Etiology and Pathology
The precise etiology of hydranencephaly is unknown, but most investigators believe compromise of the internal carotid artery circulation before 16 gestational weeks followed by diffuse liquefactive necrosis of the cerebral mantle is responsible. Maternal trauma, toxins, twin-twin transfusion syndrome, massive hemorrhage, and infection have all been cited as possible contributory factors. COL4A1 mutations with
(38-22B) The thinned calvarium has been partially removed to show the fluid-filled cavity. The hemispheres are absent ("water-bag brain"), and only the basal ganglia are present. Note separation . (Courtesy R. Hewlett, MD.)
large prenatal hemorrhages have been implicated in some cases.
In hydranencephaly, most of the cerebral hemispheres have been destroyed and are totally or partially replaced by translucent thin-walled sacs of CSF that fill most of the supratentorial space (38-22) (38-23). The outer layer consists of leptomeninges, and the inner layer is glial tissue without demonstrable ependymal elements.
The falx is intact. Cortical loss is massive but seldom complete. The medial temporal lobes, brainstem, cerebellum, and parts of the thalami—all supplied by the posterior circulation—are often relatively preserved. As some of the choroid plexus is also supplied by the posterior circulation, CSF continues to be elaborated but not normally resorbed. This distends the fluidfilled sacs that are the dominant pathologic feature of hydranencephaly.
Hydranencephaly usually occurs sporadically without other associated malformations. Fowler syndrome is a rare autosomal-recessive disorder in which hydranencephaly is accompanied by glomeruloid vasculopathy of the CNS vessels and neurogenic muscular atrophy.
Clinical Issues
Hydranencephaly occurs in 1-2 per 10,000 live births and represents 0.6% of CNS malformations in perinatal/neonatal autopsy series.
Prognosis is poor. Half of liveborn infants with hydranencephaly die within the first postnatal month, and 85% die by the end of the first year. Occasional long-term survivors have been reported. The major management problem is controlling the macrocephaly that usually
Congenital Malformations of the Skull and Brain
1238
accompanies hydranencephaly. Patients with hemihydranencephaly have a better prognosis and may experience long-term survival.
Imaging
General Features. A normal or large head with fluid-filled cranial vault ("water bag" brain) and small nubbins of remnant brain with a normal falx cerebri and posterior fossa are the typical findings (38-22) (38-23).
CT Findings. NECT scans show that CSF almost completely fills the supratentorial space. The falx cerebri is generally intact and appears to "float" in the water-filled cranial vault (38-24). The basal ganglia are present and separated but may appear moderately atrophic. Small remnants of the medial frontal and parietooccipital lobes can be present.
MR Findings. MR demonstrates a largely absent cerebral mantle. The falx is easily identified (38-25). The fluid-filled
(38-23) This is the same case as Figure 38-22, seen from above. A falx cerebriand tentorium are present, as are the separated basal ganglia. The hemispheres are absent. (Courtesy R. Hewlett, MD.) (38-24) NECT shows hydranencephaly. Both hemispheres are replaced by CSF. BG/thalami are separated , falx is present . No brain is visible over CSF-filled cavities .
(38-25A) Sagittal T1WI shows hydranencephaly with macrocephaly; CSF fills virtually all of the supratentorial spaces. Brainstem and cerebellum are normal. (38-25B) Coronal T1WI in the same case shows expanded, CSF-filled cranial vault, only tiny remnants of brain . A falx is present. (Courtesy A. Illner, MD.)
spaces follow CSF on all sequences, although some signal heterogeneity is often present secondary to CSF pulsations. In hemihydranencephaly, one hemisphere appears absent, and the CSF-filled space often displaces the falx across the midline.
Differential Diagnosis
The most important differential diagnosis of hydranencephaly
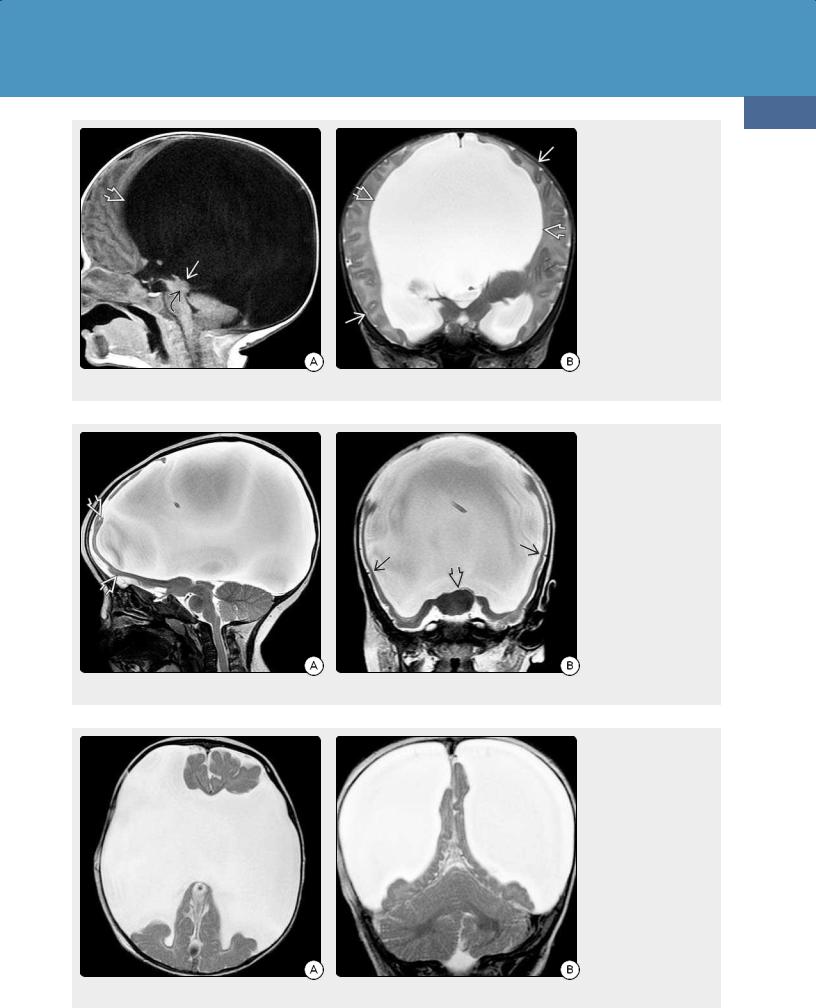
(38-25) is severe obstructive hydrocephalus (OH). In severe OH (e.g., secondary to aqueductal stenosis), a thin cortex can be seen compressed against the dura and inner table of the calvaria (38-26).
In alobar holoprosencephaly, the falx and interhemispheric fissure are absent. The basal ganglia are fused (38-27). Severe bilateral "open lip" schizencephaly has large transmantle CSF clefts that are lined with dysplastic-appearing cortex (3828). Severe cystic encephalomalacia shows large ventricles with multiple parenchymal CSF-filled cavities.
Holoprosencephalies, Related Disorders, and Mimics
1239
(38-26A) Sagittal T1WI in a 4-week infant with macrocrania shows massively enlarged lateral ventricles and tectal dysplasia causing aqueductal stenosis . (38-26B) Coronal T2WI shows the massively enlarged lateral ventricles. There is a thin rim of compressed but normally formed cortex and subcortical WM lying under the calvarium. This is maximal hydrocephalus.
(38-27A) This is alobar holoprosencephaly. Sagittal T2WI shows enlarged head with relatively normalappearing posterior fossa. Almost the entire calvarium is occupied by the CSF-filled monoventricle covered by a very thin rim of featureless brain . (3827B) Coronal T2WI shows a horseshoe-shaped monoventricle. The basal ganglia are fused. Note absent falx, thin rim of smooth dysplasticappearing brain .
(38-28A) Axial T2WI shows severe "open lip" schizencephaly, another cause of "water-bag brain" appearance. (3828B) Coronal T2WI in the same case shows that the falx and tentorium are normal. The massive "open lip" schizencephalic clefts are lined by dysplastic-appearing gray matter.

Congenital Malformations of the Skull and Brain
1240
Selected References
Holoprosencephaly
Yang E et al: A practical approach to supratentorial brain malformations: what radiologists should know. Radiol Clin North Am. 55(4):609-627, 2017
Dubourg C et al: Mutational spectrum in holoprosencephaly shows that FGF is a new major signaling pathway. Hum Mutat. 37(12):1329-1339, 2016
Gupta S et al: Roof plate mediated morphogenesis of the forebrain: new players join the game. Dev Biol. 413(2):145-52, 2016
Kaliaperumal C et al: Holoprosencephaly: antenatal and postnatal diagnosis and outcome. Childs Nerv Syst. 32(5):801-9, 2016
DeMyer W: Holoprosencephaly. In: Handbook of Clinical Neurology, Vol. 30, edited by Vinker PI, Bruyn JW. Amsterdam, The Netherlands: North Holland Publishing, 1977, pp 431-478
Alobar Holoprosencephaly
Pucciarelli V et al: Facial evaluation in holoprosencephaly. J
Craniofac Surg. 28(1):e22-e28, 2017
Rosa RFM et al: Trisomy 18 and holoprosencephaly. Am J Med
Genet A. ePub, 2017
Semilobar Holoprosencephaly
Pucciarelli V et al: Facial evaluation in holoprosencephaly. J Craniofac Surg. 28(1):e22-e28, 2017
Barkovich AJ et al: Pediatric Neuroimaging, 5e. Philadelphia, PA: Lippincott Williams & Wilkins, 2012
Lobar Holoprosencephaly
Pucciarelli V et al: Facial evaluation in holoprosencephaly. J
Craniofac Surg. 28(1):e22-e28, 2017
Holoprosencephaly Variants
Middle Interhemispheric Variant of Holoprosencephaly
Bulakbasi N et al: The middle interhemispheric variant of holoprosencephaly: magnetic resonance and diffusion tensor imaging findings. Br J Radiol. 89(1063):20160115, 2016
Virta M et al: Adult with middle interhemispheric variant of holoprosencephaly: neuropsychological, clinical, and radiological findings. Arch Clin Neuropsychol. 31(5):472-9, 2016
Yahyavi-Firouz-Abadi N et al: Case 236: middle interhemispheric variant of holoprosencephaly. Radiology. 281(3):969-974, 2016
Septopreoptic Holoprosencephaly
Esen E et al: Pyriform aperture enlargement in all aspects. J Laryngol Otol. 1-4, 2017
de Boutray M et al: Median cleft of the upper lip: a new classification to guide treatment decisions. J Craniomaxillofac Surg. 44(6):664-71, 2016
Yang S et al: Congenital nasal pyriform aperture stenosis in association with solitary median maxillary central incisor: unique radiologic features. Radiol Case Rep. 11(3):178-81, 2016
Ginat DT et al: CT and MRI of congenital nasal lesions in syndromic conditions. Pediatr Radiol. 45(7):1056-65, 2015
Poelmans S et al: Genotypic and phenotypic variation in six patients with solitary median maxillary central incisor syndrome. Am J Med Genet A. 167A(10):2451-8, 2015
Related Midline Disorders
Septooptic Dysplasia
Alt C et al: Clinical and radiologic spectrum of septo-optic dysplasia: review of 17 cases. J Child Neurol. ePub, 2017
Koizumi M et al: Endocrine status of patients with septo-optic dysplasia: fourteen Japanese cases. Clin Pediatr Endocrinol. 26(2):89-98, 2017
Ryabets-Lienhard A et al: The optic nerve hypoplasia spectrum: review of the literature and clinical guidelines. Adv Pediatr. 63(1):127-46, 2016
Arrhinencephaly
de Boutray M et al: Median cleft of the upper lip: a new classification to guide treatment decisions. J Craniomaxillofac Surg. 44(6):664-71, 2016
Kaliaperumal C et al: Holoprosencephaly: antenatal and postnatal diagnosis and outcome. Childs Nerv Syst. 32(5):801-9, 2016
Holoprosencephaly Mimics
Hydranencephaly
Adhikari EH et al: Infant outcomes among women with Zika virus infection during pregnancy: results of a large
prenatal Zika screening program. Am J Obstet Gynecol. 216(3):292.e1-292.e8, 2017
Pavone P et al: Hydranencephaly: cerebral spinal fluid instead of cerebral mantles. Ital J Pediatr. 40(1):79, 2014

Chapter 39
1241
Familial Cancer Predisposition
Syndromes
The term cancer predisposition syndrome is used to describe familial cancers in which a clear mode of inheritance can be established. The 2016 WHO classifies these as familial tumor syndromes.
The term neurocutaneous syndromes denotes a group of CNS disorders that are characterized by brain malformations or neoplasms and skin/eye lesions. These disorders have also been called phakomatoses. The term is derived from the Greek root phako, which refers to the lens; phakomatosis thus means a tumor-like condition of the eye (lens).
Most—but not all—neurocutaneous syndromes are inherited. Most—but not all—are associated with a distinct predilection to develop CNS neoplasms; these have also been called inherited cancer syndromes. Most—but again not all—of these also have characteristic cutaneous lesions. Many—but not all—also have prominent visceral and connective tissue abnormalities.
Cancer predisposition syndromes are typically uncommon, monogenic, highpenetrance disorders. Next-generation sequencing can identify patients who possess the germline genetic variants that underlie these disorders. Despite the availability of accurate genetic testing, many individuals are diagnosed on purely clinical grounds. In others, characteristic imaging features provide the first suggestion that a patient may have an inherited cancer syndrome.
In this chapter, we consider familial tumor syndromes that involve the nervous system, beginning with the neurofibromatoses and schwannomatosis. Major attention is also directed to tuberous sclerosis, von Hippel-Lindau disease, and Li-Fraumeni syndrome. We close with a brief discussion of rare familial tumor syndromes including Cowden and Turcot syndromes.
Neurofibromatosis and
Schwannomatosis
Neurofibromatosis is not a single entity but a group of genetically and clinically distinct disorders with few overlapping features. Although they are the most common CNS tumor predisposition syndromes, neurofibromatoses are multisystem disorders with both neoplastic and nonneoplastic manifestations.
Two types of neurofibromatosis are widely recognized: neurofibromatosis type 1 (NF1) and neurofibromatosis type 2 (NF2).
A third related disorder—schwannomatosis—is a rare non-NF1/NF2 syndrome characterized by multiple nonvestibular schwannomas. Together,
Neurofibromatosis and |
|
Schwannomatosis |
1241 |
Neurofibromatosis Type 1 |
1242 |
Neurofibromatosis Type 2 |
1250 |
Schwannomatosis |
1254 |
Other Common Familial Tumor |
|
Syndromes |
1254 |
Tuberous Sclerosis Complex |
1254 |
von Hippel-Lindau Disease |
1261 |
Rare Familial Cancer Syndromes |
1266 |
Li-Fraumeni Syndrome |
1267 |
Cowden Syndrome |
1269 |
Turcot Syndrome |
1270 |
Nevoid Basal Cell Carcinoma |
1271 |
Syndrome |
|
Rhabdoid Tumor Predisposition |
1272 |
Syndrome |
|
Meningioangiomatosis |
1273 |
Neurocutaneous Melanosis |
1274 |
Encephalocraniocutaneous |
1276 |
Lipomatosis |
|
Epidermal Nevus Syndrome |
1276 |
Proteus Syndrome |
1277 |
|
|
Congenital Malformations of the Skull and Brain
1242
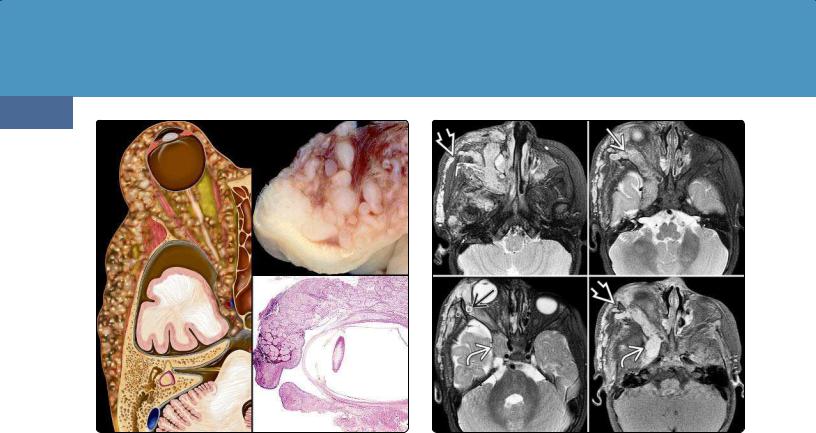
(39-1) Graphic (L) and surgical specimens (AFIP Archives) (R) depict neurofibromatosis type 1 (NF1) with typical plexiform neurofibroma of orbit, eyelid, and scalp.
these three inherited disorders affect approximately 100,000 persons in the United States alone.
Neurofibromatosis Type 1
Terminology
Neurofibromatosis type 1 (NF1) was formerly known as von Recklinghausen disease or "peripheral neurofibromatosis." Because NF1 often has central lesions, the term "peripheral neurofibromatosis" should not be used. When extreme, NF1 can be highly disfiguring and is sometimes dubbed "elephantiasis neuromatosa" or "elephant man disease."
NF1 is one of the most common of all genetic syndromes with a prevalence of 1:3,000. An uncommon form, segmental NF1 (formerly called neurofibromatosis type 5), affects one region of the body (e.g., a limb) or sometimes just a single dermatome. Segmental NF1 is a mosaicism in which localized disease results from a postzygotic NF1 gene mutation.
An even more uncommon NF1 type is localized NF1. Localized NF1 is isolated to a small area and is caused by a sporadic somatic (not germline) mutation.
Etiology
General Concepts. NF1 is an autosomal-dominant disorder with variable expression, a high rate of new mutations, and virtually 100% penetrance by age 20.
Genetics. NF1 is caused by mutation of the NF1 gene on chromosome 17q11.2. This large gene has one of the highest rates of spontaneous mutation in the entire human genome. Mutations vary from complete gene deletions to insertions,
(39-2) Series of three T2WIs, one T1 C+ FS shows enhancing plexiform neurofibroma infiltrating orbit , masticator space, and cavernous sinus . Lesion is hyperintense with typical target appearance .
stop and splicing mutations, as well as amino acid substitutions and chromosomal rearrangements.
Mutations inactivate the gene that encodes the protein product neurofibromin. Neurofibromin is a cytoplasmic protein that functions as a tumor suppressor protein through negative regulation of the RAS oncogene. Unopposed Ras activation leads to enhanced activation of the downstream RAF/MEK/ERK, PI3K/AKT/mTOR, and cAMP signaling pathways, which are critical for control of cellular growth and differentiation. NF1 is therefore considered a "Ras-opathy."
Neurofibromin also acts as a regulator of neural stem cell proliferation and differentiation; it is required for normal glial and neuronal development. The oligodendrocyte myelin glycoprotein—a major myelin protein—is also embedded in the NF1 gene and is often also mutated.
Neurofibromin is expressed at low levels in all cells with higher levels expressed in the CNS (astrocytes and oligodendrocytes as well as neurons, Schwann cells) and skin (melanocytes).
Approximately half of all NF1 cases are familial. Nearly 50% are sporadic ("de novo") and represent new mutations. NF1 patients already harboring a heterozygous germline NF1 mutation develop neurofibromas upon somatic mutation of the second (wild-type) NF1 allele. About 10% of NF1 patients display somatic mosaicism.
Pathology
CNS lesions are found in 15-20% of patients. A variety of nonneoplastic lesions as well as benign and malignant tumors are associated with NF1. An increased risk of non-CNS malignancies also occurs in NF1 patients.
Familial Cancer Predisposition Syndromes
1243
(39-3) Plexiform neurofibroma involving cervical nerve roots is depicted in the graphic (L) and on a coronal STIR scan (R).
(39-4) Coronal autopsy specimen (L) and coronal STIR (R) show plexiform neurofibroma of thoracolumbar nerve roots. (Autopsy courtesy R. Hewlett, MD.)
Nonneoplastic CNS Lesions. Multiple waxing and waning dysplastic white matter lesions on T2/FLAIR are commonly identified in patients with NF1 (see below). Histopathologically, these lesions represent zones of myelin vacuolization and dysgenesis, not hamartomas. These lesions follow a benign course with regression by 20 years.
Uncommon nonneoplastic CNS lesions include macrocephaly and subependymal glial nodules. Hydrocephalus occurs in 1015% of cases. Dural ectasia may cause dilatation of the optic nerve sheaths, Meckel cave, or internal auditory canals.
Arteriopathy occurs in at least 6% of cases. The most common manifestation is progressive intimal fibrosis of the supraclinoid internal carotid arteries, resulting in moyamoya. Both intraand extracranial aneurysms and arteriovenous fistulas occur in NF1 but are relatively rare. The vertebral arteries are more commonly affected than the carotid arteries.
CNS Neoplasms. CNS tumors occur in approximately 20% of individuals with NF1. A variety of both benign and malignant tumors occur. All involve tumorigenesis of neural crestderived cells and can be found in both the central and peripheral nervous systems. Benign NF1-related neoplasms include neurofibromas and schwannomas. Malignant tumors include malignant peripheral nerve sheath tumors and gliomas.
Neurofibromas. A spectrum of NF1-associated neurofibromas occurs. Tumors derived from skin sensory nerves are designated cutaneous or dermal neurofibromas. The prevalence of dermal neurofibromas increases with age, so more than 95% of adults with NF1 have at least one lesion.
Dermal neurofibromas are benign tumors that are composed of Schwann cells, fibroblasts, mast cells, and perineural cells. These tumors arise from a single fascicle within a peripheral nerve and appear as soft, well-circumscribed pedunculated or sessile lesions that range in size from 1 mm to 2 cm. Most patients develop more tumors as they age, and some have literally thousands of dermal neurofibromas.
Less commonly, a tumor within a larger subcutaneous nerve appears as a more diffuse mass within the dermis ("diffuse" subcutaneous neurofibroma). Neither dermal nor subcutaneous neurofibromas undergo malignant transformation.
Plexiform neurofibromas (PNFs) are distinct from dermal neurofibromas and are virtually pathognomonic of NF1. PNFs develop in 30-50% of individuals with NF1, typically manifesting at birth and growing most rapidly during the first decade of life.
PNFs are generally large bulky tumors usually associated with major nerve trunks and plexuses. PNFs are rope-like, diffusely infiltrating, noncircumscribed transspatial lesions that resemble a bag of worms (39-1).
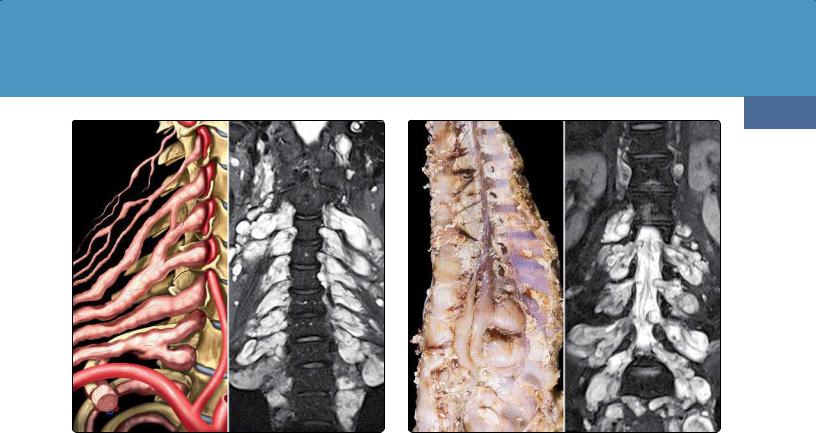
The scalp and orbit are common sites for PNFs (39-2) (39-5) (39-6). Spinal neurofibromas and PNFs are found in approximately 40% of patients with NF1 (39-3) (39-4).
Malignant Peripheral Nerve Sheath Tumors. Although most PNFs remain benign, 10-15% become malignant. Deep-seated PNFs are at particular risk for development into malignant peripheral nerve sheath tumors (MPNSTs). Individuals with NF1 have an 8-13% cumulative lifetime risk of developing an MPNST. MPNST is an aggressive, deadly tumor with a high rate of metastases and poor overall prognosis.
Congenital Malformations of the Skull and Brain
1244
(39-5) NF1 shows discrete, enhancing dermal neurofibromas , infiltrating plexiform neurofibroma , and eroding skull .
(39-6) (L) T2WI, (R) T1 C+ show scalp/orbit neurofibroma and dural dysplasia with enlarged Meckel cave .
MPNSTs may also include rhabdomyoblastic and other heterologous elements. These histologically mixed neoplasms—referred to as malignant Triton tumors—are very characteristic of NF1.
Gliomas. The overwhelming majority of CNS neoplasms in NF1 are grade I pilocytic astrocytomas (PAs). Approximately 80% of NF1 PAs arise in the optic pathway, 15% occur in the brainstem, and 5% occur in other regions.
Optic pathway gliomas (OPGs) occur in 15-20% of patients with NF1 and can be unior bilateral (39-7). Any part of the optic pathway can be involved. Some OPGs affect just the optic nerve, whereas others involve the optic chiasm and optic tracts.
NF1-associated gliomas of the medulla, tectum, and pons are typically indolent neoplasms. Approximately 20% are malignant (WHO grades II-IV). These include diffusely infiltrating ("low-grade") fibrillary astrocytoma, anaplastic astrocytoma, and glioblastoma.
Non-CNS Neoplasms. NF1 is associated with an increased risk of leukemia (especially juvenile myelomonocytic leukemia and myelodysplastic syndromes), gastrointestinal stromal tumors (6%), and adrenal or extraadrenal pheochromocytoma (0.1-5.0%).
Rare NF1-associated systemic neoplasms include rhabdomyosarcoma, juvenile xanthogranuloma, melanoma, thyroid medullary carcinoma, and glomus tumors.
NF1-ASSOCIATED NEOPLASMS
Common
•Dermal neurofibromas (95% of adults)
•Plexiform neurofibromas (PNFs) (30-50%)
•Spinal neurofibromas
Less Common
•Pilocytic astrocytoma (80% of gliomas)
○80% in optic pathway (15-20% of NF1 patients)
○15% brainstem
○5% other locations (cerebellum, cerebral hemispheres)
•Other astrocytomas (20%)
○Diffusely infiltrating fibrillary astrocytoma (WHO grade II)
○Anaplastic astrocytoma (WHO grade III)
○Glioblastoma (WHO grade IV)
Rare But Important
•Malignant peripheral nerve sheath tumor
○Develops in 8-13% of PNFs
•Juvenile chronic myeloid leukemia
•Gastrointestinal stromal tumor
•Pheochromocytoma
•Rhabdomyosarcoma
•Juvenile xanthogranuloma
•Melanoma
•Thyroid medullary carcinoma
•Glomus tumors
Clinical Issues
(39-7) Optic nerve glioma in NF1 (top), axial T2WI (bottom) show fusiform enlargement of optic nerve. Nerve sheaths are partly patulous.
Epidemiology and Demographics. NF1 is one of the most common CNS single-gene disorders, affecting 1:3,000 live births. There is no sex predilection.
Presentation. The clinical manifestations of NF1 are quite heterogeneous, and intrafamilial variation is common. Although absence of visible stigmata

Familial Cancer Predisposition Syndromes
does not exclude the diagnosis of NF1, most patients exhibit characteristic cutaneous lesions (39-8). Most are diagnosed as children or young adults.
Characteristic features include cutaneous neurofibromas (present in almost all adults with NF1), hyperpigmentary skin abnormalities with café au lait macules (95%) (39-9), inguinal/axillary freckling (65-85%), and iris hamartomas or Lisch nodules (39-10). Funduscopic examination using nearinfrared reflectance demonstrates bright patchy choroidal nodules in 70% of pediatric patients and 80% of adults.
Other less common NF1-associated features include distinctive skeletal abnormalities such as sphenoid dysplasia (3-11%), long bone deformities (1- 4%), pseudarthroses, and progressive kyphoscoliosis.
Cardiovascular anomalies occur in approximately 25% of individuals with NF1. Conotruncal cardiac defects, pulmonary valvular stenosis, and arterial hyperplasia are typical anomalies. NF1-related vasculopathy may present as renovascular hypertension, abdominal aortic coarctation, and strokes.
Cognitive impairment is common in NF1 and manifests primarily as learning difficulties and attention deficit disorder.
General surveillance recommendations in children with NF1 include annual physical examination (including ophthalmologic examination up to age 5 years), developmental assessment, and regular blood pressure monitoring. Additional specialist evaluations depend on associated CNS, skeletal, or cardiovascular manifestations.
Clinical Diagnosis. Molecular diagnostic testing distinguishes NF1 from other disorders that share similar phenotypic features. With the exception of PNF, most clinical stigmata of NF1 also occur in other disorders (e.g., multiple café au lait macules in McCune-Albright syndrome). Consensus criteria for the clinical diagnosis of NF1 are summarized in the box below.
NF1: DIAGNOSTIC CLINICAL FEATURES (AT LEAST TWO REQUIRED)
Cutaneous Lesions
•≥ 6 café au lait spots (earliest manifestation)
○Prepubertal: ≥ 0.5 cm
○Postpubertal: ≥ 1.5 cm
•Freckling of armpits or groin
•≥ 2 neurofibromas (any type)
•1 plexiform neurofibroma
Eye Abnormalities
•≥ 2 Lisch nodules (pigmented iris hamartomas)
•Optic pathway pilocytic astrocytoma
Distinctive Bone Lesion
•Sphenoid dysplasia/absence
•Long bone cortex dysplasia/thinning
Family History
• First-degree relative with NF1
Natural History. Prognosis in NF1 is variable and relates to its specific manifestations. Median age at death for all NF1 patients is 59 years. Increased mortality is related to MPNST, glioma, cardiovascular disease, and organ compression by neurofibromas.
The foci of myelin vacuolization (39-18) increase in number and size over the first decade, then regress, and eventually disappear. They are rarely identified in adults, and their relationship to intellectual impairment is uncertain.
1245
(39-8) Clinical photo reveals extensive facial plexiform neurofibroma. (Courtesy A. Ersen, MD.)
(39-9) Photographs show multiple café au lait spots (L) and cutaneous neurofibromas (R) in NF1. (Courtesy A. Ersen, MD.)
(39-10) Clinical photo shows multiple Lisch nodules in a patient with NF1. (Courtesy A. Ersen, MD.)

Congenital Malformations of the Skull and Brain
1246
(39-11A) 3D bone CT in a patient with NF1 and sphenoid dysplasia shows enlarged left orbit and widened superior orbital fissure .
(39-11B) Coronal T1 C+ FS scan in the same patient shows enhancing plexiform neurofibroma infiltrating the orbit and high deep masticator space .
Treatment Options. Foci of myelin vacuolization do not undergo neoplastic transformation and do not require treatment.
Imaging
Imaging features vary with the specific type of NF1-related abnormalities. The use of whole-body short tau inversion recovery (STIR) sequences for both identifying tumors and assessing overall tumor burden in clinical trials is becoming more widespread although standard CNS-focused sequences are illustrated here.
Nonneoplastic CNS Lesions. Bone dysplasias occur in the skull, spine, and long bones (e.g., pseudarthroses). NECT scans may demonstrate a hypoplastic sphenoid wing (39-11A) and enlarged middle cranial fossa, with or without an associated arachnoid cyst. Protrusion of the anterior temporal lobe may result in ipsilateral proptosis. The globe is frequently enlarged ("buphthalmos"), and a plexiform neurofibroma is often present (39-11B).
Dural dysplasia with patulous spinal dura (39-12) as well as enlarged optic nerve sheaths, internal auditory canals, and Meckel caves can occur (39-13).
Dysplastic white matter lesions are seen as multifocal hyperintensities on T2/FLAIR imaging. These foci represent zones of myelin vacuolization (ZMVs) and are seen in 70% of children with NF1. They generally increase in size and number until approximately 10 years of age but then wane and disappear (39-19). ZMVs are rarely seen in adults.
The most common sites are the globi pallidi (GP), centrum semiovale, cerebellar WM, dentate nuclei, thalamus, and brainstem (39-18). Most are smaller than 2 cm in diameter
(39-19). They generally exhibit little or no mass effect although the corpus callosum may appear thickened in severe cases. Confluent midbrain, tectum, brainstem, and hypothalamic lesions with unusually extensive myelin vacuolization can occasionally cause mass effect and even obstructive hydrocephalus.
Most ZMVs are isoor minimally hypointense on T1WI although GP lesions are often mildly hyperintense. ZMVs do not enhance following contrast administration and demonstrate increased ADC values on DWI.
Most NF1-associated vascular lesions are extracranial and range from renal artery stenosis to aortic coarctation and aneurysmal dilatations of the great vessels.
Relentless endothelial hyperplasia can cause progressive stenosis of the intracranial internal carotid arteries, resulting in a moyamoya pattern. Careful scrutiny of the intracranial vasculature demonstrates attenuation of the middle cerebral artery "flow voids" (39-14).
CNS Neoplasms
Neurofibromas. Patients with cutaneous neurofibromas often demonstrate solitary or multifocal discrete round or ovoid scalp lesions that are hypointense to brain on T1WI and hyperintense on T2WI. A target sign with a hyperintense rim and relatively hypointense center is common. Strong but heterogeneous enhancement following contrast administration is typical.
PNFs are most common in the orbit, where they are seen as poorly marginated serpentine masses that infiltrate the orbit, extraocular muscles, and eyelids (39-11B). They often extend
Familial Cancer Predisposition Syndromes
inferiorly into the pterygopalatine fossa and buccal spaces as well as superiorly into the adjacent scalp and masticator spaces. Transspatial extension into the neck is common. PNFs enhance strongly and resemble a "bag of worms."
Malignant Peripheral Nerve Sheath Tumors. MPNSTs arising within a PNF can be difficult to detect and to differentiate from the parent tumor. MPNSTs tend to be more heterogeneous in signal intensity, often exhibiting intratumoral cysts, perilesional edema, and peripheral enhancement. Whole-body F-18 FDG PET/CT may be helpful in distinguishing benign tumors from MPNSTs.
Gliomas. The most common glioma in NF1 is pilocytic astrocytoma. Optic pathway glioma (OPG) is the most common lesion and is seen as a diffuse, fusiform, or bulbous enlargement of one or both optic nerves (39-7). Tumor may extend posteriorly into the optic chiasm, superiorly into the hypothalamus, fornices, and cavum septi pellucidi, laterally into the temporal lobes, posteriorly into the optic tracts and
1247
lateral geniculate bodies, and posteroinferiorly into the cerebral peduncles and brainstem (39-15).
Signal intensity is variable. Most OPGs are isointense with brain on T1WI and isoto moderately hyperintense on T2WI. Enhancement on T1 C+ FS scans varies from none to striking.
MRS is generally not helpful, as pilocytic astrocytomas often demonstrate a malignant-appearing spectrum with elevated choline and increased Cho:Cr ratio. Neither extent of signal intensity nor enhancement indicates malignancy, so interval surveillance is necessary. NF1-associated OPGs can be stable for many years or involute spontaneously.
NF1-associated low-grade fibrillary astrocytomas can be difficult to distinguish from FASIs. They are usually moderately hypointense on T1WI and hyperintense on T2WI and show progression on follow-up imaging.
(39-12) Sagittal (L) and coronal (R) T2WIs show NF1 with extreme dural ectasia causing posterior vertebral scalloping and extensive meningoceles. (39-13) T2WI in NF1 shows CSF-filled patulous Meckel caves and internal auditory canals.
(39-14A) Axial T2WI in a teenager with NF1 shows extremely attenuated anterior and middle cerebral arteries , a vascular manifestation of NF1. (39-14B) Coronal T2WI in the same patient shows the typical appearance of moyamoya in NF1 with markedly attenuated supraclinoid internal carotid, anterior cerebral, and middle cerebral arteries .

Congenital Malformations of the Skull and Brain
1248
(39-15A) Axial T2WI in a patient with NF1 shows an enlarged hyperintense left optic nerve , a prosthetic globe , and a hyperintense mass in the pons. (39-15B) More cephalad T2WI shows enlarged optic chiasm , mass extending into right midbrain , and foci of abnormal signal intensity in both medial temporal lobes and the left midbrain .
(39-15C) T1 C+ FS scan shows intense enhancement in the enlarged optic chiasm , medial temporal lobes , and midbrain . (39-15D) More cephalad scan shows that the enhancement extends posteriorly along both optic radiations . Biopsy demonstrated pilocytic astrocytoma (WHO grade I) without evidence of malignant degeneration.
(39-16) Precontrast (L) and postcontrast (R) T1WIs in an 18y man with NF1 shows a circumscribed cyst with enhancing nodule. This is hemispheric pilocytic astrocytoma, WHO grade I. (39-17) (L) T1 C+ FS in a 30y woman with NF1, headaches shows a tiny enhancing focus in the left parieto-occipital lobe. The patient was lost to followup but returned 12 years later. The mass is very large, abuts the dura . This is gliosarcoma, WHO IV.
Familial Cancer Predisposition Syndromes
Anaplastic astrocytoma and glioblastoma multiforme are more aggressive, more heterogeneous tumors that demonstrate relentless progression. A progressively enlarging mass that enhances following contrast administration in a child with NF1 should raise suspicion of malignant neoplasm.
NF1: IMAGING
Scalp/Skull, Meninges, and Orbit
•Dermal neurofibromas
○Solitary/multifocal scalp nodules
○Increases with age
○Localized, well-circumscribed
•Plexiform neurofibroma
○Pathognomonic of NF1 (30-50% of cases)
○Large, bulky infiltrative transspatial lesions
○Scalp, face/neck, spine
○Orbit lesions may extend into cavernous sinus
•Sphenoid wing dysplasia
○Hypoplasia → enlarged orbital fissure
○Enlarged middle fossa ± arachnoid cyst
○Temporal lobe may protrude into orbit
•Dural ectasia
○Tortuous optic nerve sheath
○Patulous Meckel caves
○Enlarged IACs
Brain
•Hyperintense T2/FLAIR WM foci
○Wax in first decade, then wane
○Rare in adults
•Astrocytomas
○Most common: pilocytic
○Optic pathway, hypothalamus > brainstem
○Malignant astrocytoma (anaplastic astrocytoma, glioblastoma multiforme) less common
Arteries
•Progressive ICA stenosis → moyamoya
•Fusiform ectasias, arteriovenous fistulas
○Vertebral > carotid
Differential Diagnosis
In combination with appropriate clinical findings (see above), the presence of ZMVs on MR with or without OPG is diagnostic of NF1. In and of themselves, multifocal T2/FLAIR hyperintensities are nonspecific and can be seen in a variety of nonneoplastic disorders, including demyelinating disease and viral encephalitis. Unlike NF1, viral encephalitis has an acute clinical course and is usually associated with encephalopathy.
Unusually extensive, confluent FASIs can mimic neoplasm (i.e., pilocytic astrocytoma, diffusely infiltrating low-grade astrocytoma, anaplastic astrocytoma, glioblastoma multiforme, or gliomatosis cerebri). Both FASIs and gliomas are part of the NF1 spectrum, so follow-up imaging may be necessary.
A recently described disorder parallels NF1 in some ways—multiple café au lait macules, axillary freckling, and macrocephaly—but the causative gene (SPRED1) is different. This disorder has been termed NF1-like syndrome. Patients lack cutaneous neurofibromas or PNFs, typical NF1 bone lesions, and optic pathway gliomas.
1249
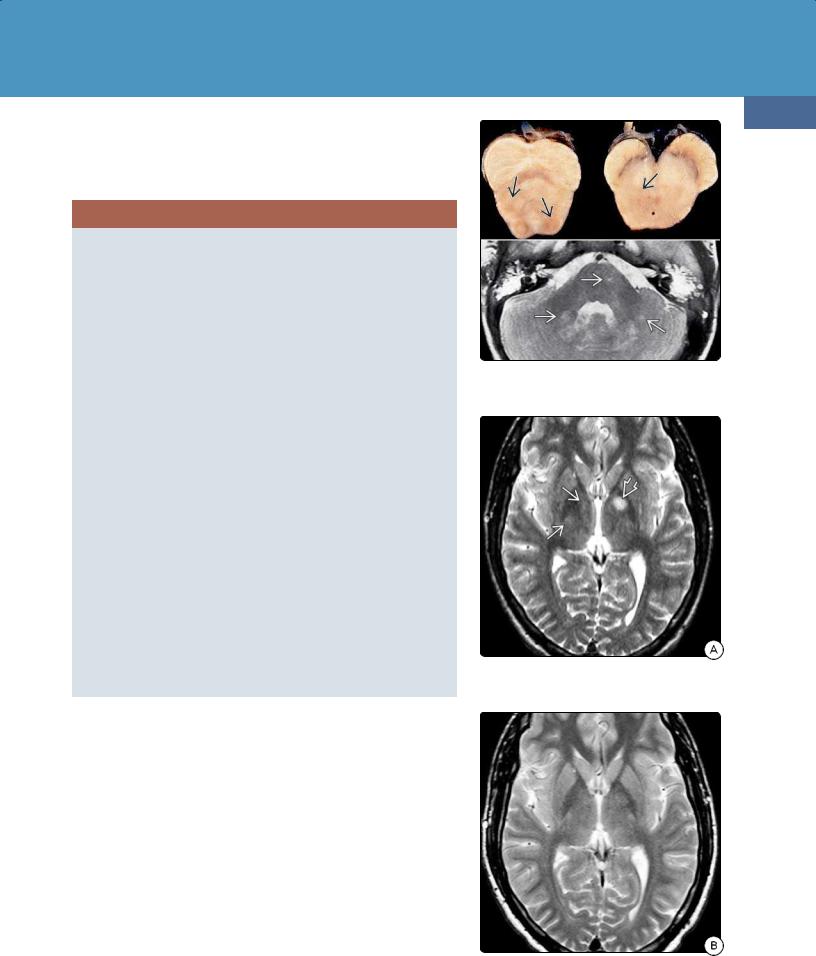
(39-18) Autopsied NF1 (top) shows foci of discolored WM (AFIP). T2WI (bottom) shows hyperintense lesions in pons, cerebellum.
(39-19A) T2WI in a child with NF1 shows zones of myelin vacuolization (ZMVs) in right and left basal ganglia .
(39-19B) Six years later, the ZMVs have resolved completely without residual abnormality.

Congenital Malformations of the Skull and Brain
1250
(39-20) Graphic depicts classic NF2 with bilateral vestibular schwannomas , facial schwannoma , and cavernous sinus meningioma .
Neurofibromatosis Type 2
Although historically grouped with NF1, neurofibromatosis type 2 (NF2) is a distinct syndrome with totally different mutations, clinical features, and imaging findings. Neurofibromas characterize NF1 and are composed of Schwann cells plus fibroblasts. Schwannomas (especially bilateral vestibular schwannomas) are the major feature of NF2 and contain only Schwann cells.
The associated neoplasms are also different from those in NF1. Astrocytomas are found in NF1, whereas ependymomas and meningiomas are the predominant tumors in NF2.
There is only one similarity between NF1 and NF2: they both predispose affected individuals to develop benign Schwann cell tumors.
Terminology
NF2 is also known as neurofibromatosis with bilateral vestibular ("acoustic") schwannomas. Historically, NF2 was termed central neurofibromatosis (to distinguish it from socalled peripheral neurofibromatosis, i.e., NF1). The term von Recklinghausen neurofibromatosis is associated only with NF1 and should not be used for NF2.
Etiology
General Concepts. Like NF1, NF2 is an autosomal-dominant disorder. About half of all cases occur in individuals with no family history of NF2 and are caused by newly acquired germline mutations. Approximately 30% of these patients have mosaic genetic alterations.
(39-21) Autopsy (top) shows bilateral vestibular schwannomas in NF2. (A. Ersen, MD.) Typical T1 C+ scan (bottom) shows bilateral vestibular and facial schwannomas and right cavernous sinus trigeminal schwannoma.
Genetics. NF2 is caused by mutations of the NF2 gene on chromosome 22q12. The NF2 gene encodes the protein Merlin (moesin-erzin-radixin-like protein), which is also known as schwannomin. Merlin is implicated in the regulation of membrane organization and cytoskeleton-based cellular processes, such as adhesion, migration, cell-cell contact, and signaling.
Merlin functions as a growth inhibitor and tumor suppressor and regulates antiangiogenic factors. Inactivating mutations of the NF2 gene result in loss of contact-dependent inhibition of proliferation and cause predominantly benign neoplasms (schwannomas and meningiomas). Biallelic NF2 inactivation is detected in the majority of sporadic meningiomas and nearly all schwannomas.
Pathology
Location. CNS lesions are present in virtually all patients with NF2.
The most common NF2-related schwannomas are vestibular schwannomas (VSs) (39-20). Approximately 50% of patients have nonvestibular schwannomas (NVSs). The most common locations for NVSs are the trigeminal and oculomotor nerves. NF2-associated schwannomas of the trochlear and lower cranial nerves occur but are relatively rare.
Meningiomas occur in approximately half of all patients with NF2 and can be found anywhere in the skull and spine. The most frequent sites are along the falx and cerebral convexities.
Intracranial ependymomas are rare in NF2. Most are found in the spinal cord, especially within the cervical cord or at the cervicomedullary junction.

Familial Cancer Predisposition Syndromes
1251
(39-22) Autopsy specimen demonstrates innumerable small meningiomas , a common finding in NF2. (From DP: Neuropathology, 2e.)
Size and Number. NF2-related schwannomas, meningiomas, and ependymomas are often multiple. The presence of bilateral VSs is pathognomonic of NF2; adult patients with NF2 have an average of three meningiomas.
Size varies from tiny to several centimeters. Innumerable tiny schwannomas ("tumorlets") throughout the cauda equina are seen in the majority of patients. Intramedullary ependymomas are often small; multiple tumors are present in nearly 60% of patients.
Gross Pathology. NF2 is characterized by multiple schwannomas, meningiomas, and ependymomas. Virtually all patients have bilateral VSs, considered the hallmark of NF2 (39-21). Most schwannomas are well-delineated round or ovoid encapsulated masses that are attached to—but do not infiltrate—their parent nerves.
Multiple meningiomas are the second pathologic hallmark of NF2 (39-22). They are found in approximately 50% of patients and may be the presenting feature (especially in children). Meningiomas appear as unencapsulated but sharply demarcated masses.
Microscopic Features. Schwannomas are composed of neoplastic Schwann cells. Areas of alternating high and low cellularity (Antoni A pattern) are admixed with foci that exhibit microcysts and myxoid changes (Antoni B pattern). Schwann cells are strongly immunoreactive for S100 and usually do not express Merlin.
Benign nerve sheath tumors, especially of the peripheral nerves, can occasionally exhibit hybrid features of both neurofibroma AND schwannoma.
(39-23) Sagittal NECT in a 31y woman with NF2, multiple intracranial and spinal schwannomas shows innumerable hyperdense calcified masses that abut the dura and falx characteristic of NF2-associated meningiomatosis.
Staging, Grading, and Classification. Although NF2associated schwannomas often have higher proliferative activity than sporadic tumors, they are not necessarily more aggressive. They are considered WHO grade I tumors.
Most NF2-associated meningiomas are WHO grade I neoplasms. Among symptomatic resected meningiomas, grades II and III tumors are found in 29% and 6% of cases, respectively. NF2-associated ependymomas—especially those in the spinal cord—are generally indolent and carry a favorable prognosis.
Clinical Issues
Epidemiology and Demographics. NF2 is much less common than NF1, with an estimated prevalence of 1:25,000 births. There is no geographic, ethnic, or sex predilection.
Presentation. Unlike NF1 patients, individuals with NF2 generally do not become symptomatic until the second to fourth decades; symptoms often precede definitive diagnosis by 5-8 years. Average age at initial diagnosis is 17-24 years; less than 20% of patients with NF2 present under the age of 15.
Unlike NF1, most of the clinical features of NF2 involve the nervous system. Cutaneous schwannomas and/or juvenile subcapsular opacities may be the first visible manifestations of NF2. Café au lait spots are seen in only one-quarter of patients and are both less prominent and fewer in number than in individuals with NF1.
Most adult patients exhibit CN VIII dysfunction with progressive sensorineural hearing loss, tinnitus, and difficulties with balance. Other common symptoms include facial pain and/or paralysis, vertigo, and seizures. Hearing loss is relatively

Congenital Malformations of the Skull and Brain
1252
(39-24A) T2WI in a 14y boy with NF2 reveals lesions in the right cavernous sinus and both internal auditory canals (IACs) .
(39-24B) More cephalad T2WI in in the same case shows a hypodense mass in the right cavernous sinus and lesions in the left CPA cistern .
(39-24C) T1 C+ FS scans show right cavernous sinus meningioma , CN III , and left CNs IV, V , and VIII schwannomas.
uncommon in children. Subcapsular cataracts, seizures, facial nerve palsy, and other cranial neuropathies are common.
Many NF2-related meningiomas are asymptomatic and discovered incidentally on imaging studies; if symptoms appear, seizures or focal neurologic deficits are the most common. Spinal cord ependymomas are asymptomatic in 75% of patients.
Clinical Diagnosis. The definitive diagnosis of NF2 is established genetically. Similar to NF1, consensus criteria have been developed for the clinical diagnosis and are summarized in the box below. Findings are divided into those of "definite" and "probable" NF2.
NF2: DIAGNOSTIC CLINICAL FEATURES
Definite NF2
•Bilateral vestibular schwannomas (VSs)
•First-degree relative with NF2 and unilateral VS diagnosed before 30 years of age
•Or first-degree relative with NF2 and 2 of the following
○Meningioma
○Glioma
○Schwannoma
○Juvenile posterior subcapsular lenticular opacities or cataracts
Probable NF2
•Unilateral VS diagnosed < 30 years of age and 1 of the following
○Meningioma
○Glioma
○Schwannoma
○Juvenile posterior subcapsular lenticular opacities or cataracts
•≥ 2 meningiomas and 1 of the following
○1 VS diagnosed < 30 years of age
○1 meningioma, glioma, schwannoma, or lens opacity
Natural History. Actuarial survival for NF2 patients after diagnosis is 85% at 5 years, 67% at 10 years, and 38% at 20 years. Although NF2-associated meningiomas have a mean annual growth rate of 1.5 mm, de novo meningiomas with brain edema may require active treatment.
NF2-associated intracranial neoplasms often demonstrate a "saltatory" growth pattern characterized by alternating periods of growth and quiescence.
As new tumors can develop and radiographic progression and symptom development are unpredictable, continued surveillance is necessary.
Treatment Options. Treatment is increasingly conservative with the use of stereotaxic radiosurgery and drugs like bevacizumab, a monoclonal antibody against VEGF. Current recommended MR surveillance includes imaging at 1, 5, and 10 years postoperatively.
Imaging
General Features. The cardinal imaging feature of NF2 is bilateral vestibular schwannomas.
CT Findings. NECT scans typically demonstrate a mass in one or both cerebellopontine angle (CPA) cisterns. Both schwannomas and meningiomas are typically isoto slightly hyperdense on NECT (39-23) and exhibit strong enhancement following contrast administration.
Nonneoplastic choroid plexus calcifications in atypical locations (e.g., temporal horn) are a rare manifestation of NF2 but can be striking. Bone CT

Familial Cancer Predisposition Syndromes
typically shows that one or both internal auditory canals are widened, whereas schwannomas of other cranial nerves may demonstrate enlargement and remodeling of their exit foramina.
MR Findings. MR findings of NF2-related schwannomas and meningiomas are similar to those of their sporadic counterparts. If NF2 is suspected on the basis of brain imaging, the entire spine and spinal cord should be screened. High-resolution T2WI and contrast-enhanced sequences disclose asymptomatic tiny schwannomas (39-24) (39-27) and intramedullary ependymomas (39-25) in at least half of all individuals with NF2 (39-26).
Dural enhancement at the porus acusticus is present in approximately 10% of extracanalicular VSs. Although it may represent dural reaction, hypervascularity, or neoplastic infiltration, it portends increased tumor adherence and greater likelihood of subtotal resection and should be considered in surgical planning.
Differential Diagnosis
The major differential diagnosis of NF2 is schwannomatosis. Schwannomatosis is characterized by multiple NVSs. Meningiomas are less common in schwannomatosis. Multiple meningiomatosis is characterized by multifocal meningiomas without schwannomas.
NF1 vs. NF2 vs. SCHWANNOMATOSIS
Neurofibromatosis Type 1
•Common (90% of all neurofibromatosis cases)
•Chromosome 17 mutations
•Almost always diagnosed by age 10
•Cutaneous/eye lesions common (> 95%)
○Café au lait spots
○Lisch nodules
○Cutaneous neurofibromas (often multiple)
○Plexiform neurofibromas (pathognomonic)
•CNS lesions less common (15-20%)
○T2/FLAIR hyperintensities (myelin vacuolization; lesions wax, then wane)
○Astrocytomas (optic pathway gliomas—usually pilocytic—other gliomas)
○Sphenoid wing, dural dysplasias
○Moyamoya
○Neurofibromas of spinal nerve roots
Neurofibromatosis Type 2
•Much less common (10% of all neurofibromatosis cases)
•Chromosome 22 mutations
•Usually diagnosed in second to fourth decades
•Cutaneous, eye lesions less prominent
○Mild/few café au lait spots
○Juvenile subcapsular opacities
•CNS lesions in 100%
○Bilateral vestibular schwannomas (almost all)
○Nonvestibular schwannomas (50%)
○Meningiomas (50%)
○Cord ependymomas (often multiple)
○Schwannomas of spinal nerve roots
Schwannomatosis
•Very rare; usually de novo mutation
•Multiple nonvestibular schwannomas ± meningioma
•SMARCB1 (INI1) and LZTR1 mutations
1253
(39-25) Axial gross pathology in NF2 shows intramedullary ependymoma with cyst expanding cervical cord. (Courtesy R. Hewlett, MD.)
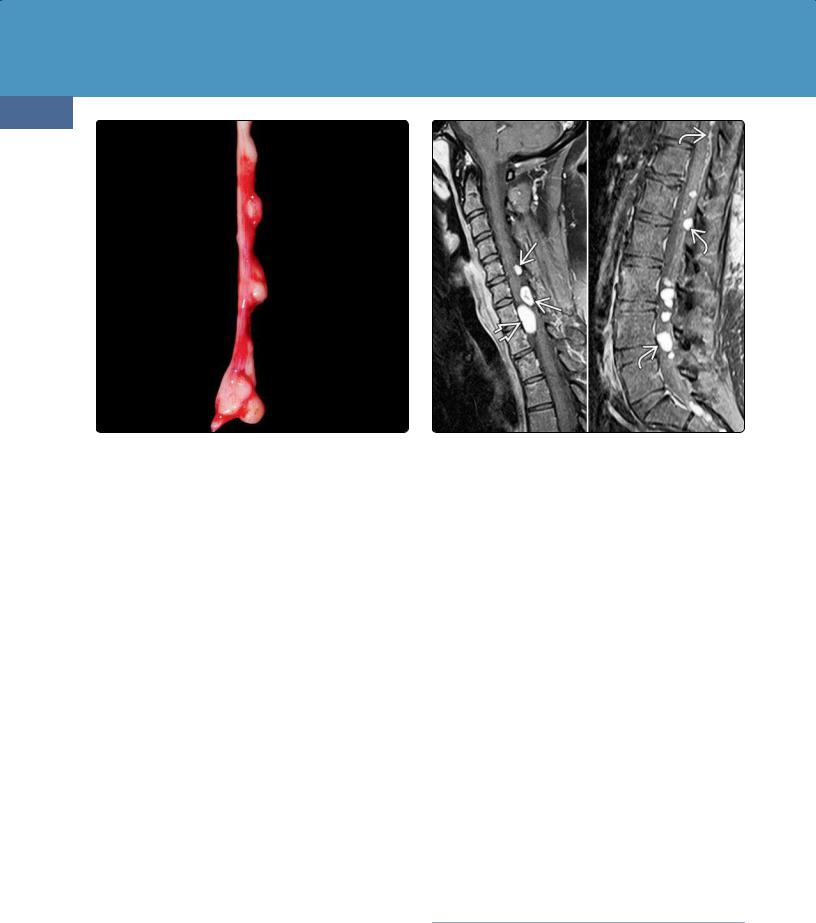
(39-26) Autopsy (L) shows intramedullary ependymomas . (Courtesy A. Ersen, MD.) (R) T1 C+ shows multiple cord ependymomas in NF2.
(39-27) NF2 graphic (L) depicts spinal "tumorlets", meningioma . T2WI (middle), T1 C+ (R) show cauda equina schwannomas.
Congenital Malformations of the Skull and Brain
1254
(39-28) Gross pathology of schwannomatosis shows multiple eccentric, expanded foci along this peripheral nerve. (From DP: Neuropathology, 2e.)
Schwannomatosis
Terminology
Schwannomatosis, which is the third major form of neurofibromatosis, is a rare hereditary cancer syndrome in which patients develop multiple nonvestibular schwannomas. Schwannomatosis is a paradigm for a tumor predisposition syndrome caused by the concomitant mutational inactivation of two or more tumor suppressor genes.
Unlike NF1 and NF2, most cases of schwannomatosis arise de novo. Less than 15% appear to be familial. The rate of transmission to offspring is low, likely due to the high rate of genetic mosaicism in founder mutations.
Etiology and Pathology
Despite the clinical overlap with NF2, schwannomatosis is not caused by germline NF2 mutations. Instead, germline mutations of either the SMARCB1 or LZTR1 tumor suppressor genes occur in 85% of familial and 40% of sporadic schwannomatosis patients. Large parts of chromosome 22q harboring, not only SMARCB1 and LZTR1, but also NF2 are affected.
Multiple schwannomas of the spine (75%), subcutaneous tissues (15%), and nonvestibular cranial nerves (10%) are characteristic (39-28). Schwannomas vary from multiple discrete nodules to plexiform lesions. Histologic features are those of typical schwannoma.
Recent evidence indicates that schwannomatosis patients with SMARCB1 mutations are at risk to develop multiple
(39-29) T1 C+ FS of (L) cervical, (R) lumbar spine in a 32y woman with arm pain, numbness show cervical meningioma , schwannomas plus innumerable cauda equina schwannomas. Intracranial scan was negative; this is schwannomatosis.
cranial meningiomas. Nearly two-thirds of meningiomas in patients with this tumor predisposition syndrome are located at the falx cerebri.
Clinical Issues
Schwannomatosis is the rarest of the neurofibromatoses, affecting 1:40,000 births. Symptoms vary, but pain is the most common presentation. Multiple painful progressive swellings in the body without the characteristic features of NF1 and NF2 should raise the suspicion of schwannomatosis. Prognosis is excellent, as anaplastic transformation is very rare.
Imaging and Differential Diagnosis
Cranial nonvestibular schwannomas are common and resemble both sporadic and NF2-associated schwannomas. Multiple enhancing nodules occur along the cauda equina and peripheral nerves. Meningiomas occur but are uncommon compared with NF2.
The major differential diagnosis of schwannomatosis is NF2. By definition, schwannomatosis lacks the bilateral VSs characteristic of NF2.
Other Common Familial
Tumor Syndromes
Tuberous Sclerosis Complex
Tuberous sclerosis complex (TSC) is a neurocutaneous syndrome characterized by the formation of nonmalignant hamartomas and neoplastic lesions in the brain, heart, skin,

Familial Cancer Predisposition Syndromes
kidney, lung, and other organs. It is associated with autism, seizures, and neurocognitive and behavioral disabilities. Because its clinical manifestations vary widely, establishing the diagnosis of TSC was particularly challenging prior to the advent of modern neuroimaging and genetic phenotyping.
Terminology
TSC has also been called Bourneville or Bourneville-Pringle disease. The classic clinical triad of TSC consists of facial lesions ("adenomata sebaceum"), seizures, and mental retardation.
Etiology
General Concepts. Approximately 50% of TSC cases are inherited and follow an autosomal-dominant pattern. The other half represents de novo mutations and germline mosaicism.
1255
Genetics. Two separate genes are mutated or deleted in TSC: TSC1 and TSC2. TSC2 mutations are approximately five times as frequent as those affecting TSC1.
The TSC1 gene is located on chromosome 9q34 and encodes a protein called hamartin. The TSC2 gene is localized to chromosome 16p13.3 and encodes the tuberin protein. Mutations in either gene are identified in 75-85% of patients with TSC.
The TSC1/TSC2 protein dimer complex functions as a tumor suppressor. Hamartin/tuberin inhibits the complex signaling pathway called mammalian target of rapamycin (mTOR). The mTOR protein product is a component of two complexes, mTORC1 and mTORC2. Activation of either mTORC regulates protein synthesis and cell growth. Mutations that lead to increased mTOR activation promote cellular disorganization, overgrowth, and abnormal differentiation that may result in tumorigenesis.
(39-30) Axial graphic of typical brain involvement in tuberous sclerosis complex (TSC) shows a giant cell astrocytoma in the left foramen of Monro, subependymal nodules , radial migration lines , and cortical/subcortical tubers. (39-31A) Autopsy specimen from a patient with TSC shows multiple expanded gyri with the potato-like appearance characteristic of cortical tubers .
(39-31B) Axial cut section from the same case shows bilateral subependymal giant cell astrocytomas and cortical tubers . (39-31C) Axial section from the same case through the lateral ventricles shows the "heaped-up" appearance of subependymal nodules along the striothalamic groove . (All three images courtesy R. Hewlett, MD.)
Congenital Malformations of the Skull and Brain
1256
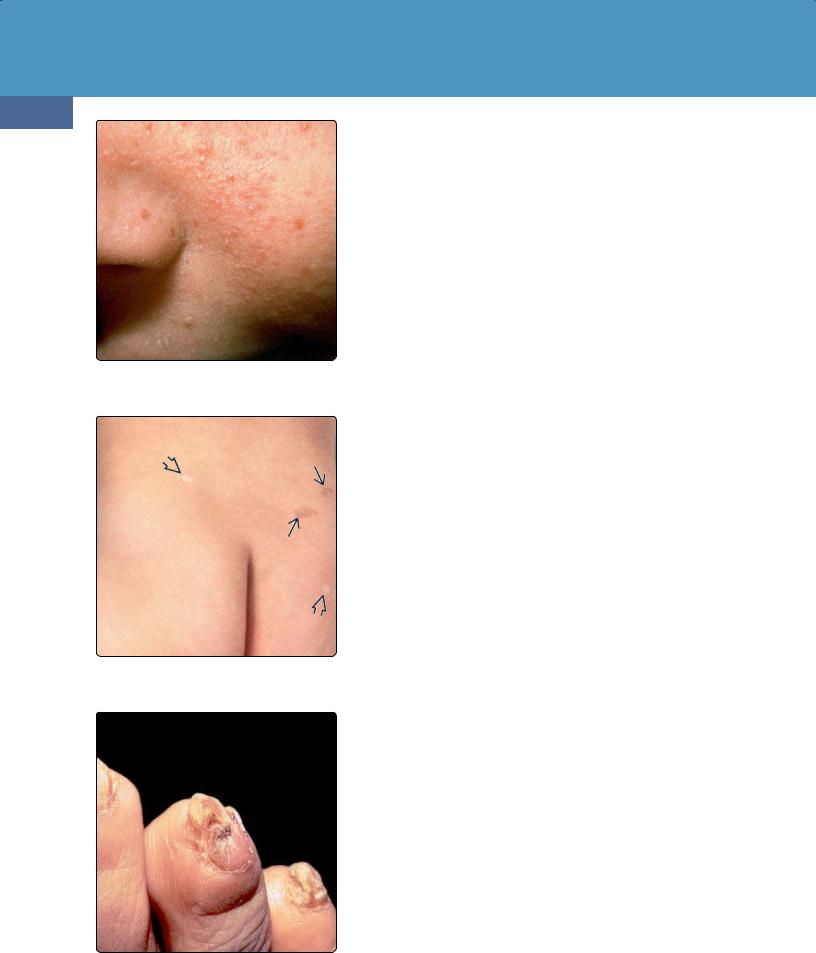
(39-32) Clinical photo shows the typical facial "adenomata sebaceum" seen in tuberous sclerosis complex. (Courtesy B. Krafchik, MD.)
(39-33) TSC is shown with "ash leaf" spots . Other macules show areas of hyperpigmentation. (Courtesy B. Krafchik, MD.)
(39-34) Periungual fibromas are common in the toes and fingernails in patients with TSC. (Courtesy B. Krafchik, MD.)
Multiple genotype-phenotype studies have demonstrated that TSC2 mutations are associated with a more severe disease phenotype with more and larger tubers, more radial migration lines, and more subependymal nodules (SENs) compared with TSC1.
Pathology
The four major pathologic features of TSC in the brain are cortical tubers, SENs, white matter (WM) lesions, and subependymal giant cell astrocytoma (39-30) (39-31).
Cortical Tubers. Cortical tubers are glioneuronal hamartomas. They are found in over 90% of TS patients and appear as firm, whitish, pyramidshaped, elevated areas of smooth gyral thickening, with or without central depressions. Cortical tubers grossly resemble potatoes ("tubers").
Microscopically, cortical tubers consist of giant cells and dysmorphic neurons with foci of gliosis, disrupted lamination, and disordered myelin. Balloon cells similar to those seen in Taylor-type focal cortical dysplasia (FCD IIb) are also commonly found in tubers. Tubers do not undergo malignant transformation.
Subependymal Nodules. SENs are located immediately beneath the ependymal lining of the lateral ventricles, along the course of the caudate nucleus.
SENs appear as elevated, rounded, hamartomatous lesions that grossly resemble candle guttering or drippings. They often calcify with increasing age. SENs along the caudothalamic groove adjacent to the foramen of Monro may undergo neoplastic transformation into subependymal giant cell astrocytoma (SEGA).
White Matter Lesions. WM lesions are almost universal in patients with TSC. They appear as foci of bizarre dysmorphic neurons and balloon cells in the subcortical WM and/or fine radial lines extending outward from the ependymal ventricular surface toward the cortex. These radial migration lines often terminate in a tuber.
Subependymal Giant Cell Astrocytoma. SEGA is seen almost exclusively in the setting of TSC, occurring in 6-9% of patients. Grossly, SEGAs appear as well-circumscribed solid intraventricular masses located near the foramen of Monro. SEGAs are WHO grade I tumors that often cause obstructive hydrocephalus but do not invade adjacent brain. Although most SEGAs are unilateral, bilateral tumors occur in 10-15% of cases.
Typical microscopic features are large (not truly giant), plump cells that resemble astrocytes and/or ganglion cells in a fibrillar background. Tumor cell GFAP positivity varies, but most SEGAs are positive for neurofilament protein, neuron-specific enolase, and synaptophysin on immunohistochemistry.
Intratumoral calcifications are relatively common, but necrosis is rare. Mitoses are few, and the MIB-1 index is generally low.
Clinical Issues
Epidemiology and Demographics. TSC is the second most common inherited tumor syndrome (after NF1) with a prevalence of approximately 1:6,000 live births. Almost 80% of cases are diagnosed before the age of 10 years. Between 20 and 30% are diagnosed during the first year of life when infantile spasms are observed in the patients with a positive family history. Patients with TSC2 mutations are diagnosed an average of 9 years earlier than patients with a TSC1 mutation.
Familial Cancer Predisposition Syndromes
Presentation. TSC patients generally present within the first two decades of life. The most common skin lesions are hypomelanotic macules, which are ovoid depigmented areas with irregular margins that are best visualized by ultraviolet light (Woods lamp). These "ash leaf" spots are seen in over 90% of cases and may be the first visible manifestation of TSC (39-33). Other common cutaneous findings such as forehead plaques, shagreen patches, facial angiofibromas ("adenoma sebaceum") (39-32), and periungual fibromas (39-34) usually do not appear until after puberty.
Clinical Diagnosis. The clinical diagnosis of TSC is problematic because all cutaneous features are age-dependent and may not become apparent until later in childhood. The classic triad of facial "adenomata sebaceum" (39-32), seizures, and mental retardation is seen in only 30% of patients.
The various clinical features of TSC are designated as either major or minor features. Based on these features, the diagnosis is divided into definite, probable, and possible TSC (see box below). Although DNA testing is useful for diagnosis and determining the causative mutation, approximately 30% of patients with definite TSC have negative results for TSC1 and TSC2 mutations.
TSC: DIAGNOSTIC CLINICAL FEATURES
Diagnosis
•Definite TSC
○2 major features or 1 major + 2 minor
•Probable TSC
○1 major + 1 minor feature
•Possible TSC
○1 major or ≥ 2 minor features
Major Features
•Identified clinically
○≥ 3 hypomelanotic ("ash leaf") macules (97%)
○Facial angiofibromas (75%) or forehead plaque (15-20%)
○Shagreen patch (45-50%)
○Ungual/periungual fibroma (15%)
○Multiple retinal hamartomas (15%)
•Identified on imaging
○Subependymal nodules (98%)
○Cortical tubers (95%)
○Cardiac rhabdomyoma (50%)
○Renal angiomyolipoma (50%)
○Subependymal giant cell astrocytoma (15%)
○Lymphangioleiomyomatosis (1-3%)
Minor Features
•Identified clinically
○Gingival fibromas (70%)
○Affected first-degree relative (50%)
○Pitting of dental enamel (30%)
○Retinal achromic patch (35%)
○Confetti-like skin macules (2-3%)
•Identified on imaging
○WM hamartomas, radial migration lines (100%)
○Hamartomatous rectal polyps (70-80%)
○Nonrenal hamartomas (40-50%)
○Bone cysts (40%)
○Renal cysts (10-20%)
Natural History. TSC is characterized by wide phenotypic variation in disease severity and natural course. Neurologic manifestations—primarily intractable seizures from brain hamartomas and obstructive hydrocephalus
1257
(39-35A) NECT in a 22y woman with TSC demonstrates typical calcifications seen in subependymal nodules.
(39-35B) NECT scan shows additional calcified SENs , wedge-shaped hypodensities characteristic of the WM lesions in TSC.
(39-35C) CECT scan shows enhancement adjacent to the foramen of Monro, suspicious for subependymal giant cell astrocytoma (SEGA).
Congenital Malformations of the Skull and Brain
1258
secondary to SEGA—are the leading cause of morbidity and mortality.
SEGAs are benign and usually slow-growing neoplasms. Although they can develop at any age, they are most frequent in patients between 5-19 years of age.
Treatment Options. Until recently, few treatment options other than surgery for SEGA existed. Rapamycin inhibitors such as everolimus and sirolimus have been approved for the treatment of TSC-associated SEGAs in patients with TSC. When growing cells are treated with rapamycin, both mTORC1 and mTORC2 are depleted. Downregulation of general protein synthesis, upregulation of macroautophagy, and activation of stress-responsive anabolic proteins occur.
Imaging
General Features. Imaging studies in TSC are abnormal in over 98% of all patients.
(39-36A) T1WI in a 42y man with TSC shows multiple hyperintense calcified SENs and SEGA in the right frontal horn. Note poorly defined gray-white matter junctions of typical cortical tubers. (39-36B) T2WI in the same case shows the hypointense calcified SENs and mixed signal intensity SEGA .
(39-36C) T1 C+ FS shows that the SEGA enhances intensely. The SENs also enhance moderately. (39-36D) T2WI (L), FLAIR (R) show cortical tubers as expanded, hyperintense gyri with poor gray-white matter differentiation , and "flame-shaped" subcortical hyperintensities . The tubers are easier to appreciate on the FLAIR image.
CT Findings
Cortical Tubers. Neonatal and infantile cortical tubers are initially seen as hypodense cortical/subcortical masses within broadened and expanded gyri (39-35B). The lucency decreases with age; tubers in older children and adults are mostly isodense with cortex.
Calcifications in cortical tubers progressively increase with age. By 10 years, 50% of affected children demonstrate one or more globular or gyriform cortical calcifications. Between 15 and 25% of all TSC patients demonstrate focal cerebellar calcifications.
Subependymal Nodules. SENs are a near-universal finding in TSC. Most are found along the caudothalamic groove. The walls of the atria and temporal horns of the lateral ventricles are less common sites.

Familial Cancer Predisposition Syndromes
SENs are rarely calcified in the first year of life. Calcification in SENs increases with age. Eventually, 50% demonstrate some degree of globular calcification (39-35A). SENs typically do not enhance on CECT scans. An enhancing or enlarging SEN—especially if located near the foramen of Monro—is suspicious for SEGA (39-35C).
White matter lesions. Most WM lesions are relatively small and difficult to detect on CT scans.
Subependymal giant cell astrocytoma. SEGAs show mixed density on NECT scans and frequently demonstrate focal calcification. Frank hemorrhage is rare. Moderate enhancement on CECT is typical.
MR Findings. In general, MR is much more sensitive than CT in depicting parenchymal abnormalities in TSC. Findings vary with lesion histopathology, patient age, and imaging sequence.
1259
Cortical tubers. In infants, tubers appear as thickened hyperintense cortex compared to the underlying unmyelinated WM on T1WI and become moderately hypointense on T2WI. "Streaky" linear or wedge-shaped T2/FLAIR hyperintense bands may extend from the tuber all the way through the WM to the ventricular ependyma (3937).
Signal intensity changes after myelin maturation. Tubers gradually become more isointense relative to cortex on T1WI (unless calcification is present and causes T1 shortening). Occasionally the outer margin of a tuber is mildly hyperintense to GM, while the subcortical component appears hypointense relative to WM (39-39).
Tubers in older children and adults demonstrate mixed signal intensity on T2/FLAIR. The periphery of the expanded gyrus is isointense with cortex while the deeper component is strikingly hyperintense. Between 3-5% of cortical tubers show mild enhancement on T1 C+ imaging.
(39-37A) T2WI in a 3y child with TSC shows linear/flame-shaped WM hyperintensities under cortical tubers . A SEGAin right frontal horn does not invade the adjacent parenchyma. (39-37B) More cephalad scan demonstrates cortical tubers and multiple radial hyperintensities extending outward from the ventricles through the corona radiata toward cortical tubers . Calcified SENs appear hypointense relative to brain.
(39-38) Axial FLAIR in a 5y boy with TSC shows a SEGA , multiple cortical tubers , and numerous CSF-like cysts in the deep periventricular white matter . (39-39) Axial FLAIR in an 11y boy with TSC shows SENs , cortical tubers , one large subcortical component , and several small deep WM CSF-like cysts .

Congenital Malformations of the Skull and Brain
1260
(39-40) Axial MIP of an SWI in a patient with TSC shows that calcified SENs "bloom" on T2* sequences.
(39-41A) Sagittal T2WI shows that a SEGA in the frontal horn is wedged into the foramen of Monro.
(39-41B) Sagittal postcontrast MP-RAGE shows that the SEGA enhances intensely but heterogeneously.
Subependymal nodules. SENs are seen as small (generally < 1.3 cm) nodular "bumps" or "candle gutterings" that protrude from the walls of the lateral ventricles (39-36). In the unmyelinated brain, SENs appear hyperintense on T1WI and hypointense on T2WI. With progressive myelination, the SENs gradually become isointense with WM.
Calcified SENs appear variably hypointense on T2WI or FLAIR (39-39) and are easily identified on T2* sequences (GRE, SWI) (39-40). They can be distinguished from blood products on the SWI phase map, as Ca++ is diamagnetic and appears bright, whereas paramagnetic substances (blood products) are hypointense.
Enhancement of SENs following contrast administration is variable. About half of all SENs show moderate or even striking enhancement, which—in contrast to enhancement on CECT—does not indicate malignancy.
SENs are stable lesions. However, as SENs near the foramen of Monro may become malignant, close interval follow-up is essential. It is the interval change in size seen on serial examinations—not the degree of enhancement—that is significant. Some investigators suggest an increase of greater than 20% demonstrated on two consecutive MR scans as defining a SEGA.
White Matter Lesions. WM lesions are seen in 100% of cases. Even though they are considered a "minor" criterion for TSC, their appearance is highly characteristic of the disease. Streaky linear or wedge-shaped lesions extend along radial bands from the ventricles to the undersurfaces of cortical tubers (39-37). In the unmyelinated brain, these linear foci (radial migration lines) appear mildly hyperintense to WM on T1WI. In older children and adults, they are hyperintense on T2/FLAIR sequences.
Small round cyst-like parenchymal lesions are seen in nearly 50% of TS cases. They are typically located in the deep periventricular white matter (39-38). They are often multiple and resemble CSF, i.e., they suppress on FLAIR and do not enhance.
Subependymal Giant Cell Astrocytoma. Although SEGAs can occur anywhere along the ventricular ependyma, the vast majority are found near the foramen of Monro. SEGAs are mixed signal intensity on both T1and T2WI (39-41). Virtually all enhance moderately strongly on T1 C+ scans (3936C).
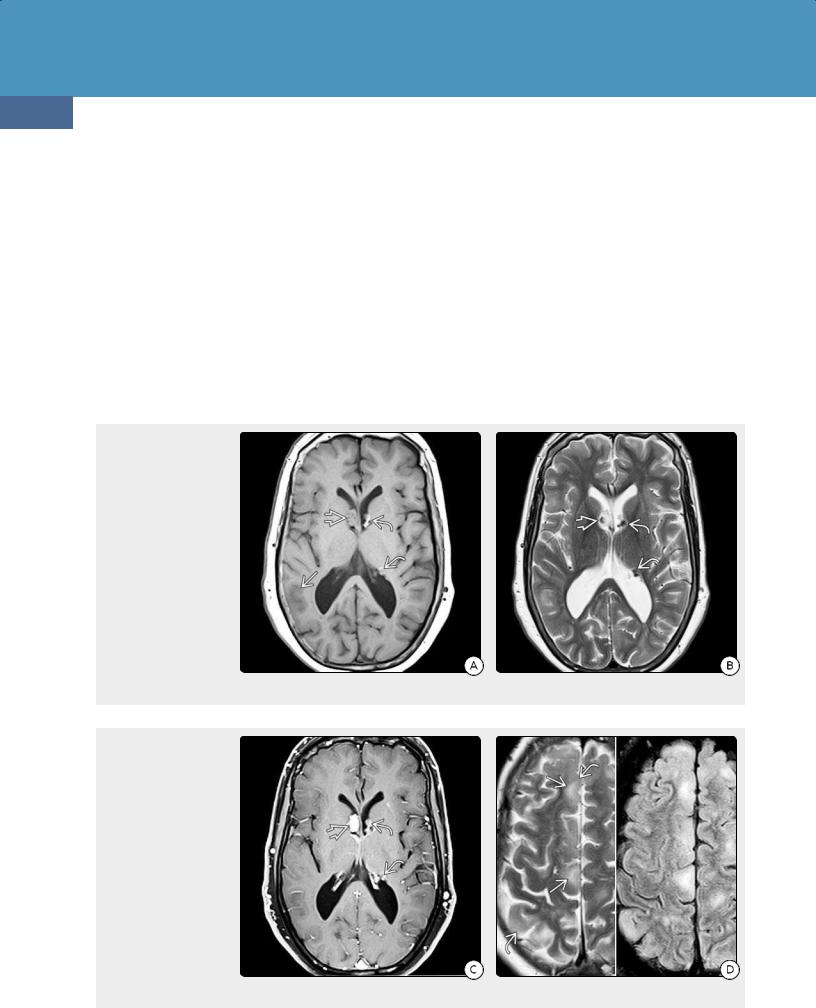
SEGAs become symptomatic when they obstruct the foramen of Monro and cause hydrocephalus. Even large SEGAs rarely invade brain.
Miscellaneous CNS Lesions. Cerebellar tubers can be identified in 10-40% of cases and are always associated with supratentorial lesions. Other uncommon abnormalities include hemimegalencephaly, cerebellar malformations, and linear, clump-like, or gyriform parenchymal calcifications. Aneurysms (mostly fusiform aortic and intracranial) are seen in 1% of TSC.
Differential Diagnosis
Brain somatic mutations in TSC1 and TSC2 can cause focal cortical dysplasia (FCD). Although FCD can appear identical on imaging studies and histopathology, lesions are typically solitary, whereas cortical tubers are almost always multiple. Foci of subependymal heterotopic gray matter can resemble SENs, but most SENs calcify and often enhance on T1 C+ sequences.
SEGAs can resemble other frontal horn/septum pellucidum lesions such as subependymoma. Subependymomas are tumors of middle-aged and older individuals, and other TSC stigmata such as cortical tubers and SENs are absent.
Familial Cancer Predisposition Syndromes
The WM T2/FLAIR hyperintense streaks of radial glial lines extend from the subventricular zone to cortical tubers. Medullary veins follow the same general course but appear hypointense on T2* SWI and enhance following contrast administration. The CSF-like white matter cysts can resemble enlarged perivascular spaces but are typically embedded within abnormalappearing WM.
TSC: IMAGING
Cortical Tubers
•Broad, expanded gyrus
•CT: initially hypodense; Ca++ increases with age
○50% of patients eventually develop ≥ 1 calcified tuber(s)
•MR: periphery isointense, subcortical portion T2/FLAIR hyperintense
Subependymal Nodules
•CT: Ca++ rare in first year, increases with age
○50% eventually calcify; don't enhance
•MR: T1 hyper-, T2 hypointense; 50% enhance
White Matter Lesions
•T2/FLAIR hyperintense radial lines/wedges
•CSF-like cysts in deep periventricular WM
Subependymal Giant Cell Astrocytoma
•CT: mixed-density mass at foramen of Monro, moderate enhancement
•MR: heterogeneous signal, strong enhancement
Miscellaneous Lesions
•Vascular (usually fusiform aneurysms), seen in 1% of cases
•Parenchymal calcifications
von Hippel-Lindau Disease
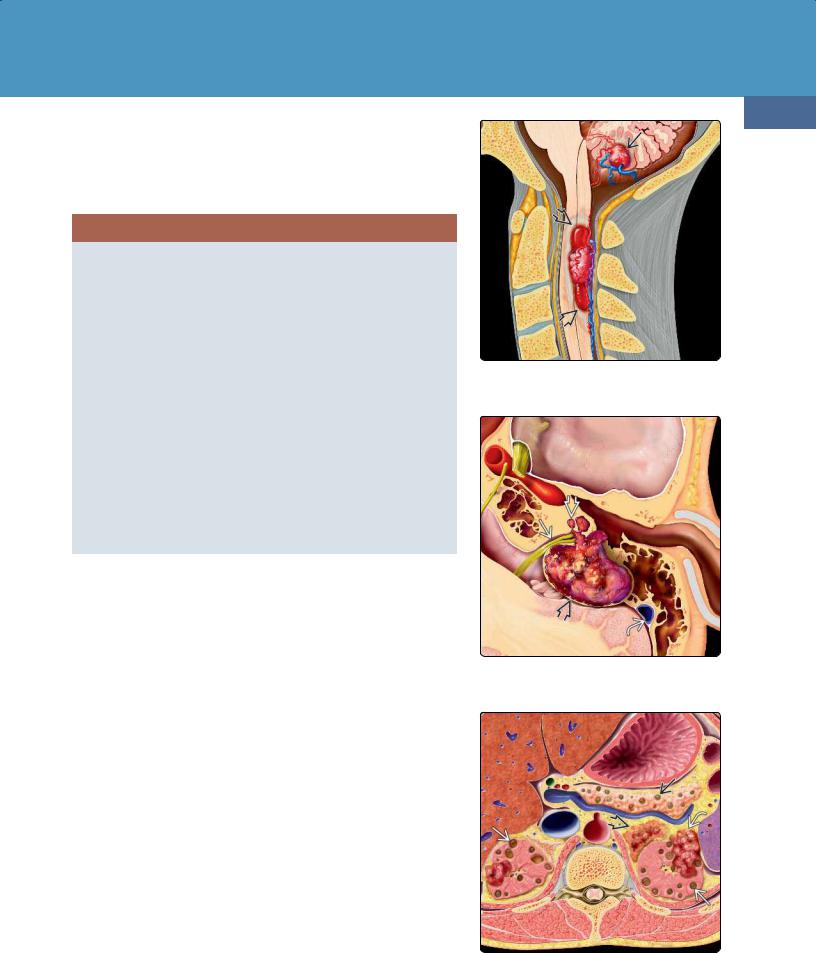
Terminology
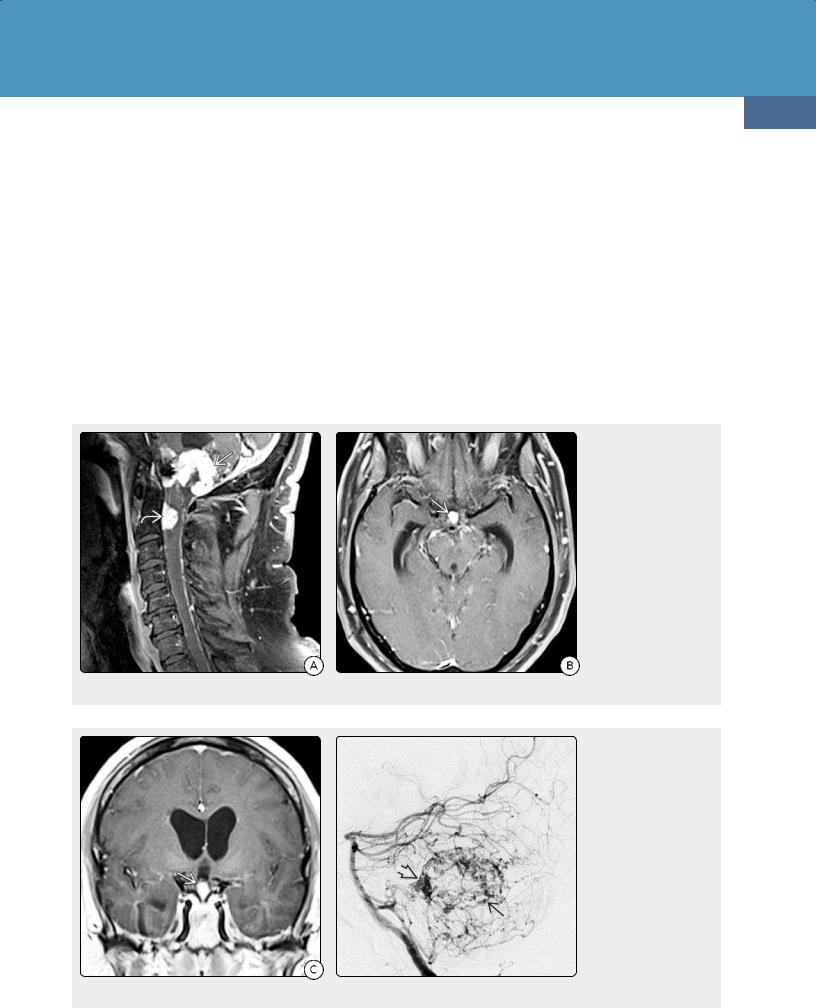
von Hippel-Lindau disease (VHL) is also known as von Hippel-Lindau syndrome and familial cerebello-retinal angiomatosis. VHL is characterized by retinal and CNS hemangioblastomas (HBs) (39-42), endolymphatic sac tumors (ELSTs) (39-43), abdominal neoplasms (adrenal pheochromocytomas, clear cell renal carcinomas), and pancreatic and renal cysts (39-44).
Etiology
General Concepts. VHL is an autosomal-dominant familial tumor syndrome with marked phenotypic variability and age-dependent penetrance.
Genetics. Mutations in the VHL tumor suppressor gene on chromosome 3p25.3 cause inactivation of the VHL protein (pVHL) and increased expression of factors such as PDGF and VEGF, which in turn leads to angiogenesis and tumorigenesis. Approximately 20% of tumors in patients with VHL result from de novo germline mutations.
Two VHL phenotypes are recognized, distinguished by the presence or absence of associated pheochromocytoma. Type 1 has a low risk of pheochromocytoma and is caused by truncating mutations of the VHL gene. Type 2 is caused by missense mutations and has a high risk of developing pheochromocytoma. Type 2 VHL is subdivided into type 2A [low risk of renal cell carcinoma (RCC), 2B (high risk of RCC), and 2C (familial pheochromocytoma without either hemangioblastoma or RCC)].
1261
(39-42) Two HBs in VHL show spinal cord tumor has associated cyst , causing myelopathy. Small cerebellar HB would be asymptomatic.
(39-43) ELST is a lytic, vascular, hemorrhagic mass between IAC and sigmoid sinus . Note tendency to fistulize inner ear .
(39-44) Abdominal VHL lesions include bilateral renal cysts , carcinomas , pancreatic cysts, and adrenal pheochromocytoma .
Congenital Malformations of the Skull and Brain
1262
VHL: GENETICS
Type 1 VHL
•Genotype = truncating mutations
•Phenotype
○Low risk for pheochromocytoma (PCC)
○Retinal angioma, CNS hemangioblastomas (HBs)
○Renal cell carcinoma (RCC), pancreatic cysts, neuroendocrine tumors
Type 2 VHL
•Genotype = missense mutation
•Phenotypes
○All have high risk of PCC
○Type 2A (low risk of RCC); retinal angiomas, CNS HBs
○Type 2B (high risk of RCC); retinal angioma, CNS HBs, pancreatic cysts, neuroendocrine tumor
○Type 2C (risk for PCC only); no HB or RCC
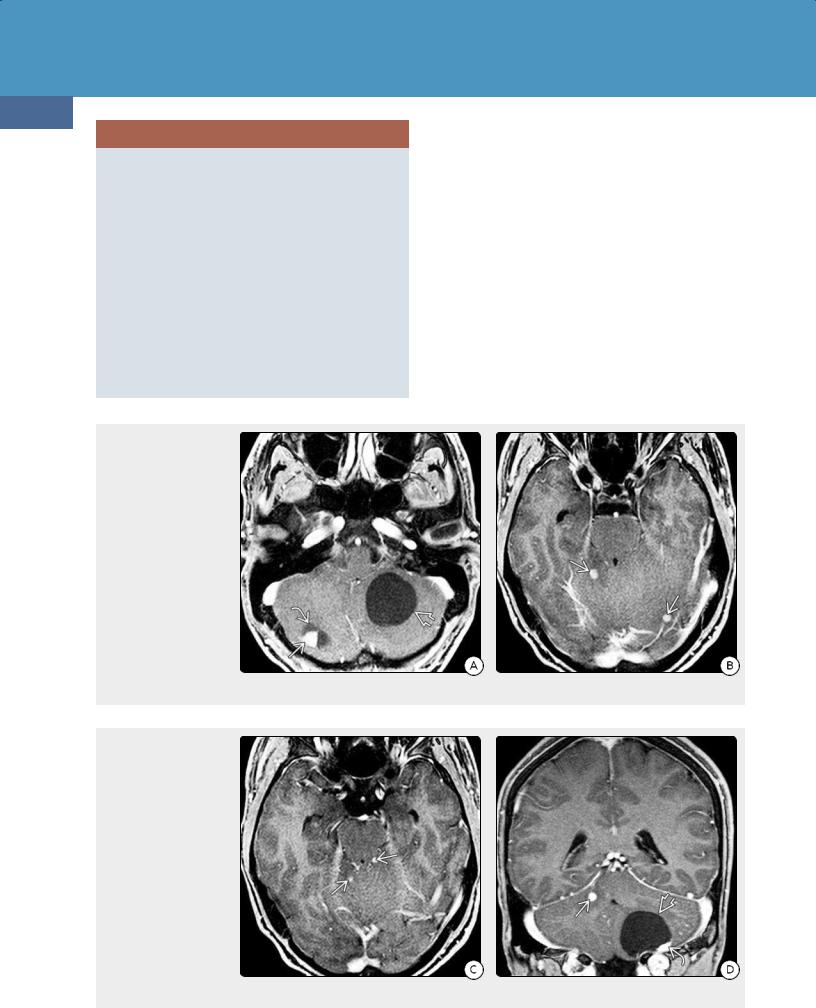
(39-45A) Axial T1 C+ FS scan in an asymptomatic 26y man with pancreatic cysts and a strong family history of VHL shows a large cystic mass in the left cerebellar hemisphereand a smaller cyst with enhancing nodule in the right hemisphere. (39-45B) More cephalad scan in the same case shows two tiny enhancing nodules .
(39-45C) Even more cephalad scan in the same patient shows another two tiny enhancing nodules in the upper cerebellum . (39-45D) Coronal T1 C+ shows the enhancing nodule associated with the large left cerebellar cyst. The nodule abuts a pial surface; the cyst wall consists of compressed, gliotic brain and does not enhance. Note separate enhancing nodule in the right hemisphere. This is classic VHL-associated hemangioblastoma.
Pathology
Many of these de novo mutations result in mosaicism. Here patients have clinical signs of the disease, but genetic testing may be negative because not all tissues carry the mutation.
The great majority of VHL patients harbor significant CNS disease. The two most common VHL-related CNS neoplasms are craniospinal HBs (found in 60-80% of all VHL cases) and ELSTs (seen in 10-15% of patients).
Hemangioblastomas. HBs are well-circumscribed red or yellowish masses that usually abut a pial surface. The vast majority of intracranial HBs are infratentorial; the dorsal half of the cerebellum is the most common site, followed by the medulla.
Approximately 10% are supratentorial; the most common site is the pituitary stalk (30% of all supratentorial HBs and 3% of those in patients with VHL). Most are asymptomatic and do
Familial Cancer Predisposition Syndromes
not require treatment. Less common locations are along the optic pathways and in the cerebral hemispheres.
Nearly half of all VHL-associated HBs occur in the spinal cord. Intraspinal HBs are often multiple and are frequently associated with a syrinx.
Between one-quarter and one-third of HBs are solid; twothirds are at least partially cystic and contain amber-colored fluid. One or more cysts together with a variably sized mural tumor nodule is the typical appearance. HBs are highly vascular with large arteries and prominent draining veins.
Two microscopic features dominate HBs and are identical in both sporadic and VHL-associated cases: a rich capillary network and large, vacuolated, variably lipid-laden stromal cells with clear cytoplasm. The cyst wall is nonneoplastic compressed brain with prominent piloid gliosis and Rosenthal fibers.
1263
HBs often demonstrate nuclear pleomorphism and hyperchromasia with scattered large, dark nuclei. Mitoses are absent or few, and MIB-1 is generally low. HBs are designated as WHO grade I neoplasms.
Retinal Hemangioblastomas ("Angiomas"). Retinal capillary angiomas are the typical ocular lesions of VHL and are seen in half of all cases. Retinal angiomas are small but often multifocal, and almost 50% are bilateral. They are identical in histopathology to CNS HBs; the differing terminology reflects ophthalmologic tradition, not histopathologic diagnosis.
Endolymphatic Sac Tumors. ELSTs are slow-growing, benign but locally aggressive papillary cystadenomatous tumors of the endolymphatic sac. Sporadic ELSTs are more common than VHL-associated tumors. Approximately 10-15% of VHL patients develop an ELST; of these, 30% are bilateral.
Grossly, ELSTs appear as vascular "heaped up" tumors along the posterior aspect of the petrous temporal bone.
(39-46A) Sagittal T1 C+ scan in a 38y man with VHL shows multiple HBs in the cerebellum and cervical spinal cord . (39-46B) Axial T1 C+ FS scan in the same patient demonstrates an enlarged enhancing pituitary stalk.
(39-46C) Coronal T1 C+ scan shows that the pituitary stalk is enlarged and enhances intensely. The patient's endocrine status was normal. The pituitary infundibulum is the most common site for supratentorial HBs in VHL. (39-47) Lateral DSA shows typical angiographic findings in HBs. The lesions exhibit mass effect, intense vascular "stain" , and striking neovascularity with tortuous, irregularappearing vessels .

Congenital Malformations of the Skull and Brain
1264
(39-48) (L) Intraoperative photograph shows typical dorsal subpial location of HB nodule and prominent vessels . (R) Sagittal T1 C+ shows multiple HBs .
Microscopically, ELSTs demonstrate interdigitating papillary processes embedded in sheets of dense fibrous tissue. Cystic foci and evidence of old and recent hemorrhage are common.
VHL: PATHOLOGY
CNS Neoplasms
•HBs (60-80%)
○Retinal HBs ("angiomas") (50%)
•Endolymphatic sac tumors (10-15%)
Visceral Lesions
•Renal lesions (2/3 of all VHL patients)
○Cysts (50-75%)
○Clear cell renal carcinomas (25-45%)
•Adrenal pheochromocytoma (10-20%)
○Hallmark of type 2 VHL
•Pancreatic cysts (35-70%), nonsecretory islet cell tumors (5-10%)
•Epididymal cysts, cystadenomas (60% of male patients, often bilateral)
•Broad ligament cystadenomas (female patients, rare)
Clinical Issues
Epidemiology and Demographics. VHL is uncommon; estimated incidence is 1:35,000-50,000 live births.
Presentation and Clinical Diagnosis. Because all VHLassociated lesions can also occur as sporadic (i.e., nonfamilial) events, a clinical diagnosis of VHL disease in a patient without a positive family history requires the presence of at least two tumors (see box below).
(39-49) (Top) Funduscopic examination in VHL shows peripheral retinal angioma supplied by prominent arteries . (From Imaging in Neurology.) (Bottom) T1 C+ FS shows enhancing angioma with retinal detachment in VHL.
Age at diagnosis varies. Although VHL can present in children and even infants, most patients become symptomatic as young adults. Painless visual loss from retinal angiomainduced hemorrhage is often the first symptom (mean: 25 years).
Tumors are the presenting feature in approximately 40% of cases. HBs, pheochromocytomas, and endolymphatic tumors typically become symptomatic in the 30s, whereas RCCs tend to present somewhat later. Mean age at diagnosis of symptomatic RCCs is 40 years, but asymptomatic tumors are frequently detected earlier on screening abdominal CT.
VHL: DIAGNOSTIC CLINICAL FEATURES
No Family History of VHL
•≥ 2 CNS HBs or
•1 CNS HB + visceral tumor
Positive Family History of VHL
•1 CNS HB or
•Pheochromocytoma or
•Clear cell renal carcinoma
Natural History. VHL-associated HBs demonstrate a "saltatory" growth pattern characterized by quiescent periods (averaging slightly over 2 years) interspersed with periods of growth. Nearly half of all patients develop de novo lesions after the initial diagnosis of VHL.
The two major causes of death in VHL patients are RCC (50%) and HBs. Overall median life expectancy is 49 years.
Surveillance Recommendations. Imaging is crucial in the identification and surveillance of extra-CNS lesions.

Familial Cancer Predisposition Syndromes
1265
(39-50A) NECT in a 51y patient with known VHL, sensorineural hearing loss shows classic feature of VHL: hyperdense V-shaped hemorrhagic retinal detachment caused by underlying "angioma" (retinal HB). (39-50B) Temporal bone CT in same patient shows a lytic infiltrative lesion along left posterior petrous temporal bone. Note preserved "spicules" of bone within lesion. Location between the IAC, sigmoid sinus is characteristic for ELST.
(39-50C) Axial T1WI in the same patient shows that the lesion is mixed isoand hyperintense relative to brain. (39-50D) The lesion is heterogeneously hyperintense on T2WI. Note that the left vitreous body is hypointense compared with the normal right side.
(39-50E) Axial T1 C+ FS scan in the same patient shows that the lesion enhances intensely but heterogeneously. Note hyperintense retinal hemorrhage . (39-50F) Lateral selective external carotid angiogram shows that the enlarged posterior auricular arterysupplies the highly vascular tumor . This is classic endolymphatic sac tumor in VHL. (All six images courtesy D. Shatzkes, MD.)

Congenital Malformations of the Skull and Brain
1266
Identification of RCC is especially important because it is the major malignant neoplasm of VHL and one of the leading causes of mortality.
Patients with a family history of VHL should undergo annual screening (ophthalmoscopy, physical/neurologic examination) beginning in infancy or early childhood. Brain MRs are recommended every 1-3 years starting in adolescence. Abdominal MR or ultrasound screening for RCC and pancreatic tumors is recommended annually, beginning at age 16.
Methods for pheochromocytoma screening vary. Blood pressure should be monitored and 24-hour urine catecholamines obtained annually. More intense surveillance beginning at age 8 years should be considered in families at high-risk for pheochromocytoma (i.e., type 2 VHL).
Treatment Options. Laser treatment for angioma-induced retinal hemorrhages is common. Surgical resection of HBs is generally based on symptoms, not evidence of radiologic progression.
Imaging
General Features. The best imaging clue for VHL is the presence of two or more CNS HBs (39-45) or one HB plus a visceral lesion or concomitant presence of retinal hemorrhage (highly suggestive of intraocular HB).
Hemangioblastomas. HB-associated peritumoral cysts are common and often underlie neurologic morbidity and mortality. Approximately two-thirds of HBs are cystic; onethird are solid or mixed solid/cystic lesions. NECT scans typically demonstrate a hypodense cyst with isodense mural nodule that abuts the pial surface of the cerebellum. The tumor nodule enhances intensely on CECT.
MR shows that the cyst is slightly to moderately hyperintense to CSF on T1WI and isoto hyperintense on T2/FLAIR. Signal intensity of the nodule is variable; large lesions may show prominent "flow voids." Hemorrhage is common, and peritumoral edema varies.
Tumor nodules enhance strongly on T1 C+ (39-46). Enhanced scans often demonstrate several tiny nodules in the cerebellum and/or spinal cord (39-48). Less commonly, HBs are identified in the pituitary stalk (the most common supratentorial site) (39-46B) (39-46C), optic tracts, or cerebral hemispheres. An uncommon manifestation of recurrent VHL-associated HB, disseminated leptomeningeal hemangioblastomatosis, is seen as multiple tumor nodules with diffuse pial enhancement of the spinal cord and/or brain.
DSA demonstrates one or more intensely vascular masses with prolonged tumor "blush" and variable arteriovenous shunting (39-47).
Retinal Hemangioblastomas ("Angiomas"). Retinal angiomas (actually small capillary HBs) are usually visualized as hemorrhagic retinal detachments that are hyperdense compared with normal vitreous on NECT. Tiny enhancing nodules can sometimes be identified on T1 C+ MR (39-49).
Endolymphatic Sac Tumors. ELSTs are located along the posterior petrous temporal bone between the internal auditory canal and the sigmoid sinus. The imaging hallmark of ELST is that of a retrolabyrinthine mass associated with osseous erosion. Bone CT shows an infiltrative, poorly circumscribed, lytic lesion with central intratumoral bone spicules (39-50).
MR demonstrates T1 hyperintense foci in 80% of cases. Signal intensity is mixed hyperand hypointense on T2WI. Heterogeneous enhancement is seen following contrast administration. ELSTs are vascular lesions that may demonstrate prominent "flow voids" on MR and prolonged tumor "blush" on DSA (39-50F).
VHL: IMAGING
Multiple Hemangioblastomas
•Diagnostic of VHL
•2/3 cystic, 1/3 solid
•Nodule abuts pia
•50% in cord (dorsal > ventral surface)
○Multiple tiny "tumorlets" along cord common
○Disseminated leptomeningeal hemangioblastomatosis
Retinal "Angiomas"
•Hemorrhagic retinal detachment
○V-shaped hyperdense posterior globe
○Plus other HB = highly suggestive of VHL
•With or without enhancing "dots" (tiny HBs)
Unior Bilateral Endolymphatic Sac Tumors
•Dorsal T-bone
○Between IAC, sigmoid sinus
•Infiltrative, lytic, intratumoral bone spicules
•T1 iso-/hyperintense; T2 hyperintense
•Strong enhancement
Differential Diagnosis
The major differential diagnosis of VHL in the brain is sporadic non-VHL-associated hemangioblastoma. Between 60-80% of HBs are sporadic tumors not associated with VHL. Multiple HBs and/or supratentorial lesions are highly suggestive of VHL.
A pilocytic astrocytoma with cyst and mural tumor nodule can resemble a solitary HB. Pilocytic astrocytomas are solitary tumors of children, whereas HBs are rarely seen in patients younger than 15 years. In contrast to HB, the tumor nodule in pilocytic astrocytoma typically does not abut a pial surface.
Vascular metastases can mimic multiple HBs but are rarely isolated to the cerebellum and/or spinal cord.
Rare Familial Cancer
Syndromes
A number of other neurocutaneous syndromes have been identified in recent years.
Familial Cancer Predisposition Syndromes
As with the more common inherited cancer syndromes discussed above, the neoplasms associated with these disorders do not differ much—if at all—from their sporadic counterparts. They are histopathologically identical and often feature the same genetic mutations. What sets them apart is the constellation of clinical features—often skin lesions—combined with systemic and CNS neoplasms.
We close the chapter with a brief consideration of these interesting syndromes.
Li-Fraumeni Syndrome
Terminology
Li-Fraumeni syndrome (LFS) is also known as the sarcoma family syndrome of Li and Fraumeni. LFS is an autosomaldominant familial tumor syndrome characterized by a lifelong increased risk of developing multiple malignant tumors in a variety of organ systems, including the brain.
1267
Etiology
Nearly three-quarters of all patients have loss-of-function germline mutations in the tumor suppressor gene TP53 (sometimes termed the "guardian of the genome"). Two newly discovered exons of the gene, 9β and 9γ, are the targets of inactivating mutation events in breast, liver, and head and neck tumors.
Pathology
CNS Tumors. Nearly half of all CNS neoplasms in LFS are astrocytomas, primarily the diffusely infiltrating fibrillary type (WHO grades II-IV) and gliosarcoma (39-51) (39-52).
Choroid plexus tumors (15%) (39-53) and medulloblastoma/primitive neuroectodermal tumor (PNET) (10-12%) are the next most common types. Ependymoma, oligodendroglioma, and meningioma together account for less than 5% of LFS-associated neoplasms. Histologically, LFS-
(39-51A) T2WI from a surveillance MR in a 16y girl with Li-Fraumeni and proven TP53 mutation shows an infiltrating hyperintense mass in the right hippocampus and posteromedial temporal lobe. (39-51B) More cephalad T2WI shows an infiltrating hyperintense mass in the right insula, hippocampus . The mass did not enhance on T1 C+. Biopsy disclosed diffusely infiltrating astrocytoma, WHO grade II.
(39-52A) (L) Axial T2WI and (R) T1 C+ scans in a 22y man with Li-Fraumeni show surgically proven glioblastoma . (39-52B) Six months following resection, a new durabased lesion —a gliosarcoma—has developed.

Congenital Malformations of the Skull and Brain
1268
(39-53A) Coronal T2WI in a child with Li-Fraumeni syndrome shows a mass filling and slightly expanding the body of the left lateral ventricle.
associated neoplasms are indistinguishable from their sporadic counterparts.
Extraneural Manifestations. Together with brain tumors, breast cancer and sarcomas (osteosarcomas and soft tissue tumors) account for almost 75% of LFS-associated neoplasms. Other reported LFS-associated neoplasms with TP53 mutations include hematopoietic and lymphoid tumors, lung cancer, skin cancers, stomach cancer, and ovarian cancer.
Clinical Issues
The revised diagnostic clinical criteria for LFS are shown in the accompanying box.
Prognosis is poor; almost half of all patients develop an invasive cancer by age 30 and have a lifelong increased risk of osteosarcoma, soft tissue sarcoma, leukemia, breast cancer, brain tumors, melanoma, and adrenal cortical tumors. Age at tumor onset in LFS patients is significantly younger compared with their sporadic counterparts.
Individuals with LFS have a 50% chance of developing cancer by age 40. Lifetime risk approaches 75% in male patients and almost 100% in female patients, as breast cancer is the most common LFS-associated malignancy. Adrenocortical carcinoma associated with TP53 germline mutation develops almost exclusively in children.
(39-53B) Coronal T1 C+ scan in the same patient shows that the mass enhances intensely and quite uniformly. Final histopathologic diagnosis was choroid plexus carcinoma.
LI-FRAUMENI SYNDROME: DIAGNOSTIC CLINICAL FEATURES
Patient With
•Sarcoma diagnosed < 45 years of age and
•Firstor second-degree relative with
○Any cancer diagnosed < 45 years or
○Any sarcoma at any age
Or With
•Multiple tumors (exception = breast)
○2 of which are known LFS-associated neoplasms and
○The first of which occurred < age 45 years or
○Adrenocortical carcinoma or choroid plexus tumor
Imaging
Brain imaging in LFS patients with CNS symptoms varies with tumor type. Imaging findings and differential diagnoses in LFSassociated CNS neoplasms are similar to those of their sporadic counterparts.
Differential Diagnosis
The differential diagnosis of LFS is sporadic neoplasms. Imaging findings are similar, so definitive diagnosis requires clinical correlation and genetic analysis.

Familial Cancer Predisposition Syndromes
Cowden Syndrome
Terminology
Cowden syndrome (CS) is also known as multiple hamartoma-neoplasia syndrome and phosphate and tensin homolog (PTEN) hamartoma tumor syndrome.
CS involves hamartomatous overgrowth of tissues of all three embryonic origins. The characteristic hamartomas of CS are noncancerous lesions of the skin, mucous membranes, and gastrointestinal tract. The classic brain hamartoma is dysplastic gangliocytoma of the cerebellum, also known as Lhermitte-Duclos disease (LDD). If CS and LDD occur together, the disorder is known as COLD (Cowden-Lhermitte-Duclos syndrome).
CS is a multisystem disease that carries an increased lifetime risk of developing certain cancers as well as benign neoplasms. Breast, thyroid, endometrial, colorectal, and renal cancers are the major systemic carcinomas associated with CS.
Etiology
CS is an autosomal-dominant disorder with variable expression and agerelated penetrance. Over 80% of patients have germline mutation of the PTEN tumor suppressor gene with downstream abnormalities in the mTOR signaling pathways involved in cell proliferation, cell cycle progression, and apoptosis.
Pathology
The most common brain imaging finding is nonspecific macrocephaly, with or without foci of heterotopic GM (39-54) (39-55).
The most characteristic associated lesion is dysplastic cerebellar gangliocytoma (39-56) (see Chapter 19). Grossly, marked enlargement of the cerebellar hemisphere/vermis is present. The folia are enlarged and distorted but not obliterated. Macrocephaly with heterotopic GM foci is common. Microscopic features include accumulation of abnormal ganglion cells in the inner granule cell layer, loss of Purkinje cells in the middle layer, and thickening with hypermyelination of the outer (molecular) layer.
CS patients are also at increased risk of meningiomas and glial tumors, including glioblastoma and gliosarcoma.
Clinical Issues
Epidemiology and Demographics. CS is uncommon; its estimated incidence is 1:200,000-250,000. Age at onset is variable. Most cases have been identified in adults, but LDD can occur in infants.
Presentation. In addition to multiple tumors and hamartomas, CS patients may also have megalencephaly, heterotopic GM, hydrocephalus, mental retardation, and seizures. Other features include gastrointestinal polyps as well as various benign breast, thyroid, and uterine lesions.
Characteristic mucocutaneous lesions (trichilemmomas, acral keratoses, papillomatous lesions) are present in 99% of CS patients by the third decade of life.
Clinical Diagnosis. CS is diagnosed clinically by the presence of pathognomonic lesions or a combination of major and minor criteria (see box below). The pathognomonic criteria are those features most likely to be associated with CS, whereas the major and minor criteria are not as specific.
1269
(39-54) 3D CT in a 16m boy with skin lesions characteristic of Cowden syndrome (CS) and PTEN mutation shows gross macrocephaly.
(39-55) Sagittal T1WI in a 6y boy with CS and macrocephaly shows increased craniofacial proportion and prominent frontal bossing .
(39-56) Axial T2WI shows dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease) with thickened, striated cerebellar folia.
Congenital Malformations of the Skull and Brain
1270
(39-57) FLAIR scan in type 1 Turcot shows heterogeneously hyperintense left frontal mass, anaplastic astrocytoma. (Courtesy T. Tihan, MD.)
(39-58) FLAIR scan in type 2 Turcot shows typical appearance of medulloblastoma . (Courtesy T. Tihan, MD.)
(39-59) Surgical colon specimen from a patient with type 2 Turcot shows innumerable small polyps. (Courtesy T. Tihan, MD.)
COWDEN SYNDROME: DIAGNOSTIC CLINICAL FEATURES
Pathognomonic Criteria
•Dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease) and
•Characteristic mucocutaneous lesions
○Papillomatous lesions
○Facial trichilemmomas
○Acral keratoses
Major Criteria
•Breast cancer
•Thyroid cancer (especially follicular)
•Endometrial cancer
•Macrocephaly
○Occipitofrontal circumference > 97th percentile
Minor Criteria
•Other thyroid lesions
•Mental retardation
•GI polyps
•Fibrocystic breast disease
•Lipomas
•Fibromas
•Genitourinary tumors (especially renal cell carcinoma)
•Genitourinary structural malformations
•Uterine fibroids
Natural History. CS carries a significantly increased lifetime risk of developing malignancies. Women with CS have a 50% lifetime risk of developing breast cancer, 10% risk for follicular thyroid cancer, and 5-10% risk of developing endometrial cancer. The risk is even higher for patients with germline PTEN mutations and includes renal and colorectal cancers as well as melanoma.
Imaging
Imaging findings of dysplastic cerebellar gangliocytoma in the setting of CS/PTEN hamartoma tumor syndrome are identical to those of LDD. An enlarged cerebellar hemisphere with thickened folia in a "corduroy" or "tigerstriped" appearance is typical.
Differential Diagnosis
LDD plus CS is diagnostic of COLD. Patients with LDD should be screened for CS and vice versa. A dysplastic cerebellar gangliocytoma without a characteristic mucocutaneous lesion or other criteria (e.g., breast cancer, thyroid lesions) is simply Lhermitte-Duclos disease.
Turcot Syndrome
Terminology
Turcot syndrome (TS) is a rare autosomal-dominant disorder characterized by primary CNS neoplasms and two different forms of colorectal polyps.
In type 1 Turcot (TS1) (hereditary nonpolyposis colorectal cancer, HNPCC, also known as Lynch syndrome), colorectal, endometrial, gastric, pancreaticobiliary, and genitourinary tumors occur together with malignant astrocytomas.
In type 2 Turcot, colorectal tumors (familial adenomatous polyposis, FAP) and skin lesions (such as epidermoid cysts) occur together with medulloblastoma and craniofacial exostosis (39-57) (39-58) (39-59).
Familial Cancer Predisposition Syndromes
Etiology
TS1 (HNPCC) is associated with biallelic DNA mismatch repair mutations. In type 2 Turcot, mutations in the APC gene are present. APC is a large protein complex in the WNT signaling pathway controlling cell proliferation and differentiation. CNS tumors in FAP patients are typically the WNT-subtype medulloblastoma.
Pathology
Three CNS neoplasms—medulloblastoma, anaplastic astrocytoma, and glioblastoma—account for 95% of all Turcot-associated brain tumors. The histologic features of these tumors are indistinguishable from those of their sporadic counterparts.
Clinical Issues
Patients with type 1 Turcot-associated anaplastic astrocytoma or glioblastoma present earlier than those with nonsyndromic tumors. Median age at onset is 18 years. Family history of polyposis is absent.
Patients with type 2 Turcot-associated medulloblastoma present later than patients in the general embryonal tumor population. Median age is 15 years. Patients with type 2 Turcot frequently have a family history of polyposis.
Imaging
Imaging findings in Turcot-related CNS neoplasms are identical to those of their nonsyndromic counterparts.
Nevoid Basal Cell Carcinoma Syndrome
Terminology
Nevoid basal cell carcinoma syndrome (NBCCS) is also known as Gorlin or Gorlin-Goltz syndrome. Patients with NBCCS exhibit a broad spectrum of neurodevelopmental disorders and are predisposed to develop multiorgan benign and malignant neoplasms.
Etiology
NBCCS is an autosomal-dominant disease with full penetrance but variable clinical phenotypes. NBCCS is caused by germline mutations of the PTCH1 gene on chromosome 9q22. This gene encodes a glycoprotein that is a binding site for the SHH gene.
Pathology
The CNS neoplasm most commonly associated with NBCCS is medulloblastoma. Most are desmoplastic medulloblastomas, SHH subtype.
Systemic NBCCS-associated lesions include basal cell carcinomas and epidermal cysts, multiple keratocystic odontogenic tumors (KOTs) (3960), and skeletal anomalies (e.g., bifid ribs).
Clinical Issues
Epidemiology and Demographics. NBCCS has a prevalence of 1:57,000 in population-based studies.
Presentation. Patients are usually diagnosed by age 5-10 years. Basal cell carcinomas are the most common initial presentation and may appear as early as 2 years of age. Multiple enlarging jaw masses are frequent and can be asymptomatic or painful.
1271
(39-60) NBCCS graphic shows classic appearance of multiple odontogenic keratocysts . Lesions splay teeth roots and displace nerves.
(39-61A) Bone CT in a 9y boy with NBCCS shows multiple lytic lesions in the maxilla and mandible. These are typical odontogenic keratocysts.
(39-61B) Coronal bone CT in the same patient nicely demonstrates the expansile, lobulated nature of the cysts .
Congenital Malformations of the Skull and Brain
1272
(39-62A) Axial NECT scan in a patient with NBCCS demonstrates dense lamellar calcifications along the tentorium cerebelli.
(39-62B) More cephalad NECT in the same patient shows dense calcification at the tentorial apex and along the falx cerebri .
(39-62C) Scan through the corona radiata shows that the falx is thickly and densely calcified .
Medulloblastomas develop in 4-20% of patients and present within the first 2 years of life, typically with symptoms of obstructive hydrocephalus.
Natural History. The major morbidity and mortality in NBCCS is caused by its associated neoplasms. Basal carcinomas often become more aggressive after puberty and may exhibit distant metastases. KOTs eventually develop in 80% of patients and have a high recurrence rate.
Imaging
Typical brain/head and neck abnormalities seen in NBCCS are multiple jaw cysts, macrocephaly, dense lamellar dural calcifications, and medulloblastoma.
Keratocystic Odontogenic Tumors. KOTs are seen as multiple expansile, well-corticated cysts in the mandible and maxilla on bone CT. They can be unior multilocular and often exhibit scalloped borders (39-61). They typically do not enhance on CECT scans.
KOTs exhibit low to intermediate signal intensity on T1WI and appear heterogeneously hyperintense on T2WI.
Dural Calcifications. Abnormal dural calcification eventually develops in 80% of patients older than 20 years (39-62). Subtle flecks of calcium deposition along the falx can occur in very young children and—together with the unusually early appearance of medulloblastoma—should suggest the diagnosis of NBCCS.
Thick, slightly irregular lamellar calcifications along the falx, tentorium, petroclinoid ligaments, and diaphragma sellae are typical findings in teenagers and adults with NBCCS.
Medulloblastoma. The imaging appearance of NBCCS-associated medulloblastoma is identical to that of nonsyndromic tumors. Although SHH-subtype desmoplastic MBs can occur anywhere in the posterior fossa, the lateral cerebellum is the most characteristic location.
Differential Diagnosis
The major differential diagnosis of the abnormal calcifications in NBCCS is physiologic or metabolic-related dural calcifications (e.g., as occurs with secondary hyperparathyroidism). Physiologic calcification is much less striking and is rarely seen in young children. Thick dural calcifications can be seen in patients with chronic renal failure and long-term hemodialysis.
The differential diagnosis of KOTs includes periapical (radicular) cysts—usually unilocular and associated with dental caries—and dentigerous (follicular) cysts. Dentigerous cysts are seen as a single unilocular cyst surrounding the crown of an unerupted tooth.
The differential diagnoses of NBCCS-related medulloblastoma are sporadic (nonsyndromic) medulloblastoma and atypical teratoid/rhabdoid tumor. As imaging findings of the tumors are identical, look for other differentiating features, such as atypical dural calcifications and jaw cysts.
Rhabdoid Tumor Predisposition Syndrome
Rhabdoid tumor predisposition syndrome (RTPS) is characterized by markedly increased risk of developing malignant rhabdoid tumors. Most cases are caused by biallelic germline mutations with inactivation of the SMARCB1 (INI1/SNF5) tumor suppressor gene on chromosome 22q11.
The most common CNS tumor in RTPS is atypical teratoid/rhabdoid tumor
(AT/RT). AT/RT is composed of poorly differentiated neuroectodermal and

Familial Cancer Predisposition Syndromes
mesenchymal elements. The rhabdoid component can be subtle, making the diagnosis difficult.
Approximately 60% of AT/RTs are found in the posterior fossa. Tumors tend to be large bulky masses with mixed cystic and solid components that demonstrate variable enhancement following contrast administration (3963). Dissemination at the time of initial diagnosis is common.
Other familial CNS tumors associated with RTPS include choroid plexus carcinoma, which has the same SMARCB1 inactivating mutation.
The most common non-CNS neoplasm in RTPS is malignant rhabdoid tumor (MRT) of the kidney. Prognosis is poor; MRTs are highly aggressive cancers that occur in young children and are generally lethal within months or a few years.
Meningioangiomatosis
Terminology
Meningioangiomatosis (MA) is a rare neurocutaneous syndrome characterized by a focal hamartomatous lesion that involves the pia and underlying cerebral cortex.
Etiology and Pathology
Although its precise etiology is unknown, MA is a benign, slow-growing lesion of presumed hamartomatous or developmental origin. MA can occur as a solitary or multifocal lesion.
Grossly, MA appears as a reddish gyriform mass that infiltrates the pia and underlying brain. MA can occur without or with an accompanying neoplasm (most commonly meningioma).
Microscopic features are those of a plaque-like intracortical and leptomeningeal proliferation consisting of small blood vessels, meningothelial cells, and fibroblasts. The adjacent cortex may show dense gliosis and neurofibrillary tangles. MIB-1 index is low.
Clinical Issues
MA can occur sporadically or as a forme fruste of neurofibromatosis type 2 (NF2). Sporadic MA usually occurs as a single lesion in a child or young adult who presents with seizures or persistent headaches.
Imaging
NECT scans typically demonstrate an isoto slightly hyperdense cortically based lesion with nodular, linear, or gyriform calcification. The frontal or temporal lobes are the most common sites. Mass effect is minimal or absent. Little or no enhancement is seen on CECT.
MA is isoto hypointense on T1WI. Although signal intensity on T2WI varies, most MAs are moderately hypointense to adjacent brain with variable amounts of associated edema or gliosis.
T1 C+ shows mild to moderate serpentine enhancement extending over the surface of adjacent gyri and into adjacent sulci. In some cases, cortical infiltration along the penetrating perivascular spaces thickens the cortex and obliterates the normal gray-white matter interface.
A few cases of MA with associated focal cortical dysplasia have been reported.
1273
(39-63A) T1WI in a 4m infant with vomiting and bulging fontanelle shows a large, heterogeneous posterior fossa mass .
(39-63B) The mass enhances strongly but heterogeneously and has large necroticappearing nonenhancing components.
(39-63C) The solid, enhancing portion of the mass shows moderate diffusion restriction . This is AT/RT.
Congenital Malformations of the Skull and Brain
1274
(39-64) (L) MA shows thickened cortex invaded by vascular-appearing tissue . (AFIP Archives.) (R) NECT shows Ca++ in MA (different case).
(39-65) (L) T1WI (same case as above CT) shows subtle effacement of posterior medial sulci with hyperintensity on FLAIR (R).
(39-66) Axial (L), sagittal (R) T1 C+ FS scans in the same case show sulcal enhancement . This is presumed meningioangiomatosis.
Differential Diagnosis
The most important differential diagnosis of MA is neoplasm. Imaging findings are not pathognomonic, so the definitive diagnosis is generally a pathologic one. It is extremely important that the neuropathologist does not mistake MA for an invasive atypical or malignant meningioma! MA does not recur after complete resection and does not require radiation or adjuvant chemotherapy.
MENINGIOANGIOMATOSIS
Pathology
•Benign mass of proliferating meningothelial cells, small vessels
•Curvilinear plaque-like lesion
•May show focal brain invasion
Clinical Issues
• Forme fruste of NF2
Imaging
•Iso-/hypointense on T1WI; usually hypointense on T2WI
•Serpentine enhancement ± perivascular space invasion
Differential Diagnosis
• Invasive neoplasm
Neurocutaneous Melanosis
Terminology
Neurocutaneous melanosis (NCM) is a rare nonfamilial syndrome characterized by a single giant and/or multiple congenital melanocytic nevi, excessive proliferation of melanin-containing cells in the leptomeninges, and benign and malignant tumors of the CNS (39-67).
Etiology
Scattered melanocytes are normally present in the pia over the convexities and around the base of the brain, ventral brainstem, and parts of the spinal cord. Focal or diffuse proliferation of these melanin-producing cells in the skin and meninges results in NCM.
Melanocytes are derived from neural crest cells (NCCs). Around 8-10 weeks of gestation, NCC-derived pluripotent precursors migrate to the fetal epidermis via the paraspinal ganglia and peripheral nerve sheath, ultimately generating differentiated melanocytes.
NCM is thought to be a neurocristopathy caused by neural crest aberration during early embryonic development. Some abnormalities in neural tubederived cells also occur, possibly resulting in NCM-associated brain malformation (e.g., Dandy-Walker malformation) (39-69).
Pathology
CNS disease can be parenchymal or leptomeningeal, benign or malignant. Melanosis consists of focal collections of histologically benign melanotic cells. A malignant melanoma consists of proliferating anaplastic melanotic cells. The estimated prevalence of malignant melanoma in the setting of NCM is 40-60%.
Grossly, leptomeningeal melanosis appears as superficial dark gray or black pigmentation in the pia (39-69). The most common locations for parenchymal melanotic deposits are the amygdala and cerebellum, followed by the pons, thalami, and inferior frontal lobes.

Familial Cancer Predisposition Syndromes
Clinical Issues
Between 60-70% of patients with NCM develop symptoms, usually before 5 years of age. Seizures and signs of elevated intracranial pressure can occur both with leptomeningeal melanosis and malignant melanoma. The prognosis in symptomatic NCM is extremely poor.
Imaging
Unior bilateral round or ovoid T1 hyperintensities in the anterior temporal lobes are the most characteristic findings. Focal or diffuse T1 shortening in the leptomeninges with serpentine enhancement on T1 C+ scans is much less common and is generally seen only in cases in which melanotic deposits have undergone malignant transformation (39-68).
Hydrocephalus is common. Cortical invasion along the penetrating perivascular spaces may cause significant mass effect and edema.
1275
Between 8 and 10% of patients with NCM harbor an associated Dandy-Walker malformation.
NEUROCUTANEOUS MELANOSIS
Pathology
•Black or grayish deposits
•Leptomeninges, amygdala, cerebellum
•Benign or malignant melanotic cells
Imaging
•T1 hyperintensities
○Round/ovoid deposits in amygdala
○Unior bilateral
•Serpentine pial lesions, perivascular space invasion in malignant
•Variable enhancement
•8-10% Dandy-Walker malformation
(39-67) Graphic shows localized dark (melanotic) pigmentation of the leptomeninges. Inset demonstrates extension of melanosis into the brain substance along the Virchow-Robin spaces . (39-68) Axial T1 C+ MR in a patient with extensive neurocutaneous melanosis shows diffuse piasubarachnoid space enhancement. (Courtesy M. Martin, MD.)
(39-69) Autopsy of NCM shows ovoid melanotic deposits in right , leftamygdalae. Black discoloration in sylvian fissures and over the cerebellum represents diffuse leptomeningeal melanin deposits. Note Dandy-Walker malformation seen in 8-10% of NCM. (Courtesy R. Hewlett, MD.) (39-70) Axial (L), sagittal (R) T1WIs in a child with NCM show the characteristic ovoid T1 hyperintense melanotic deposit in the amygdala .
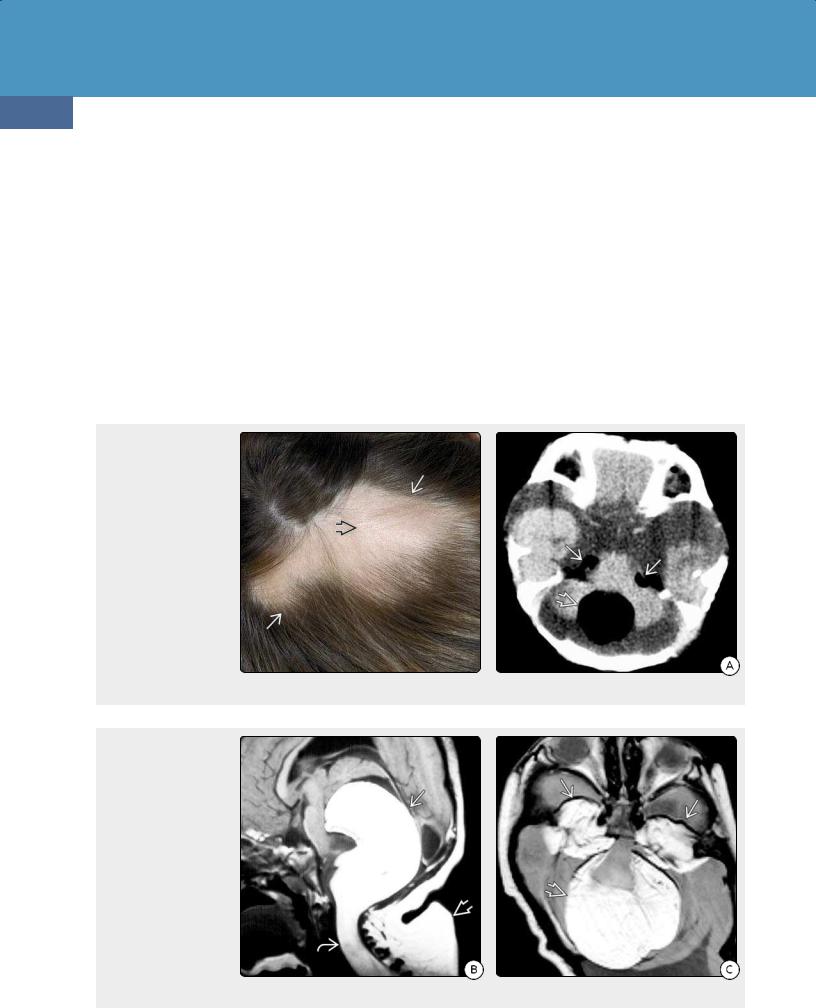
Congenital Malformations of the Skull and Brain
1276
Encephalocraniocutaneous Lipomatosis
Encephalocraniocutaneous lipomatosis (ECCL), also known as Haberland or Fishman syndrome, is a rare congenital neurocutaneous disorder whose hallmark CNS lesions are benign lipomas of the brain and spinal cord. All reported cases of ECCL are sporadic. A nonhereditary, autosomal mutation that may survive only in a mosaic state may be the cause of the clinical manifestations of ECCL.
ECCL is characterized clinically by ocular choristomas (typically lipodermoids), a smooth hairless scalp lipoma called a nevus psiloliparus (39-71), and subcutaneous cervicofacial fatty soft tissue masses. Approximately one-half of all patients have seizures, and one-third demonstrate mild or moderate mental retardation.
(39-71) Clinical photograph demonstrates a nevus psiloliparus, the dermatologic token of ECCL. A focal area of alopecia (hair loss) covers an underlying scalp lipoma , seen here as a rubbery, slightly elevated mass. (Courtesy A. Illner, MD.) (39-72A) NECT scan in a 2y child with ECCL shows focal lipomas in both cerebellopontine angle (CPA) cisterns and the cisterna magna.
(39-72B) Three years later, sagittal T1WI shows a very large suboccipital lipoma . The cisterna magna lipoma has massively increased in size. It now occupies almost the entire posterior fossa and extends inferiorly into the upper cervical spinal canal. (39-72C) Axial PD shows the large posterior fossa lipoma . The CPA lipomas have also increased in size, now extending into both Meckel caves .
Most patients with ECCL have one or more CNS lipomas. ECCL-associated lipomas have a predilection for the posterior fossa and spine. They are generally stable but may increase with age (39-72), becoming moderately large and extending over multiple spinal segments. Other congenital anomalies of the meninges such as arachnoid cysts and meningioangiomatosis are common.
Epidermal Nevus Syndrome
Epidermal nevus syndrome consists of an epidermal nevus (EN)—a benign congenital skin hamartoma—with developmental abnormalities of the skin, eyes, and CNS with variable involvement of the musculoskeletal, cardiovascular, and urogenital systems (39-73).
Several different types of nevi are included as part of epidermal nevus syndrome. The main component can be keratinocytic, sebaceous, follicular, apocrine, or eccrine. Pigmented hairy nevi, nevus comedonicus, inflammatory
Familial Cancer Predisposition Syndromes
1277
(39-73) (L) Epidermal nevus (EN) is a warty band of hypopigmented growth. (Courtesy University of Utah Department of Dermatology.) (R) EN shows hyperkeratosis, papillomatosis, and acanthosis. (Courtesy J. Comstock, MD.)
linear verrucous epidermal nevus, and linear sebaceous nevi are common manifestations.
ENs are caused by genetic mosaicism (represented by two or more different but coexisting clones of the same cell line). Embryonic epidermal cells appear early in fetal development. They proliferate and then migrate from their origin in neural crest to their destinations along so-called Blaschko lines. Mutated cells are phenotypically manifested along this pathway, resulting in the characteristic distribution of ENs.
ENs are present at birth or develop during the first few years of life. Clinically, an epidermal nevus is seen as a linear or zosteriform warty plaque that may exhibit scaly discoloration. Most are found on the neck, trunk, and extremities.
CNS malformations are present in the majority of patients with EN syndrome. The most common are malformations of cortical development (hemimegalencephaly, pachygyriapolymicrogyria) (39-74). Eye lesions are present in 40-70% of patients and include ocular colobomas, choristomas (epibulbar dermoids and lipodermoids), and optic nerve dysplasia.
Proteus Syndrome
Proteus syndrome (PS) is a rare hamartomatous disorder with multiple and diverse somatic manifestations. Mosaicism for activating mutations in AKT1 causes PS. AKT is part of the mTORC1 signaling pathway, so PS shares some genetic features with other disorders such as PTEN hamartoma tumor syndrome, TSC1/TSC2, and PIK3CA-related overgrowth spectrum.
(39-74) Axial T2WI shows right hemisphere hemimegalencephaly, the most common CNS malformation seen in epidermal nevus syndrome.
PS is characterized by localized, progressive, postnatal limb overgrowth with bony distortion, dysregulated adipose tissue, epidermal nevi, and CNS malformations. Hemimegalencephaly, pachygyria-polymicrogyria, and heterotopic gray matter are common associated abnormalities.
CNS TUMORS IN RARE FAMILIAL CANCER SYNDROMES
Li-Fraumeni Syndrome
•Most have TP53 mutations
○TP53 = "guardian of the genome"
•Astrocytoma, choroid plexus neoplasms, medulloblastoma
Cowden (MHAM) Syndrome
•PTEN mutations
•With Lhermitte-Duclos = COLD syndrome
Turcot Syndrome
•Type 1 = HNPCC (Lynch syndrome); type 2 = FAP
•Type 1 = AA, GBM; type 2 = WNT medulloblastoma
Nevoid Basal Cell Carcinoma (Gorlin) Syndrome
•PTCH1 mutations
•Medulloblastoma (usually SHH-subtype)
Rhabdoid Tumor Predisposition Syndrome
•SMARCB1/INI1/SNFS mutations
•AT/RT
Neurocutaneous Melanosis
• Leptomeningeal melanoma