

- •Pineal Parenchymal Tumors
- •Germ Cell Tumors
- •Selected References
- •Medulloblastoma
- •Selected References
- •Anatomy of the Cranial Meninges
- •Meningomas
- •Primary Melanocytic Lesions
- •Other Related Neoplasms
- •Selected References
- •Cranial Nerve Anatomy
- •Schwannomas
- •Neurofibromas
- •Selected References
- •Histiocytic Tumors
- •Selected References
- •Sellar Region Anatomy
- •Normal Imaging Variants
- •Congenital Lesions
- •Neoplasms
- •Miscellaneous Lesions
- •Selected References
- •Intracranial Pseudotumors
- •Selected References
- •Metastatic Lesions
- •Paraneoplastic Syndromes
- •Selected References
- •Scalp Cysts
- •Extraaxial Cysts
- •Parenchymal Cysts
- •Intraventricular Cysts
- •Selected References
- •Anatomy and Physiology of the Basal Ganglia and Thalami
- •Selected References
- •Alcohol and Related Disorders
- •Opioids and Derivatives
- •Inhaled Gases and Toxins
- •Selected References
- •Selected References
- •Hypertensive Encephalopathies
- •Glucose Disorders
- •Thyroid Disorders
- •Seizures and Related Disorders
- •Miscellaneous Disorders
- •Selected References
- •The Normal Aging Brain
- •Dementias
- •Degenerative Disorders
- •Selected References
- •Normal Variants
- •Hydrocephalus
- •CSF Leaks and Sequelae
- •Selected References
- •Cerebral Hemisphere Formation
- •Imaging Approach to Brain Malformations
- •Posterior Fossa Anatomy
- •Chiari Malformations
- •Hindbrain Malformations
- •Selected References
- •Commissural Anomalies
- •Malformations Secondary to Abnormal Postmigrational Development
- •Selected References
- •Anencephaly
- •Holoprosencephaly
- •Holoprosencephaly Variants
- •Related Midline Disorders
- •Holoprosencephaly Mimics
- •Selected References
- •Selected References
- •Selected References
- •Cephaloceles
- •Craniosynostoses
- •Meningeal Anomalies
- •Selected References
- •Index

Opioids and Derivatives
The ten drugs most frequently involved in overdose deaths include several opioids: heroin, oxycodone, methadone, morphine, hydrocodone, and fentanyl. Of these, heroin is the most commonly abused opioid.
In addition to the direct effects of opioids on the brain, impurities and additives may produce systemic pathology. Hypotension and anoxia may also complicate the clinical and imaging appearance of opioid toxicity.
Heroin
Heroin use has a direct and damaging effect on certain brain functions that are associated with impulsive and unhealthy decision making.
Heroin is usually injected intravenously. The most common acute complication of injected heroin is stroke. Globus pallidus ischemia, very similar to that seen in carbon monoxide poisoning, is common.
The most dramatic acute effects occur with inhaled heroin. The freebase form is heated over aluminum foil and the vapors inhaled ("chasing the dragon"). Heroin vapor inhalation causes a striking toxic leukoencephalopathy.
The most frequent secondary complication of heroin abuse is infection. Endocarditis is common and may result in septic emboli, brain abscesses, and vasculitis with mycotic aneurysm formation.
Etiology
Heroin causes both acute and chronic effects such as vasculopathy, leukoencephalopathy, and generalized brain volume loss. Stimulation of opioid receptors in vascular smooth muscle may cause reversible vasospasm. Immune-mediated response to additives in injected heroin may cause ischemia or vasculitis.
Pathology
Autopsied brains of patients with heroin-associated encephalopathy show a sponge-like appearance of the cerebral white matter. Microscopy demonstrates demyelination and white matter vacuolization. Because the cerebellum has a high density of opioid receptors, similar changes can be seen in its white matter.
Imaging
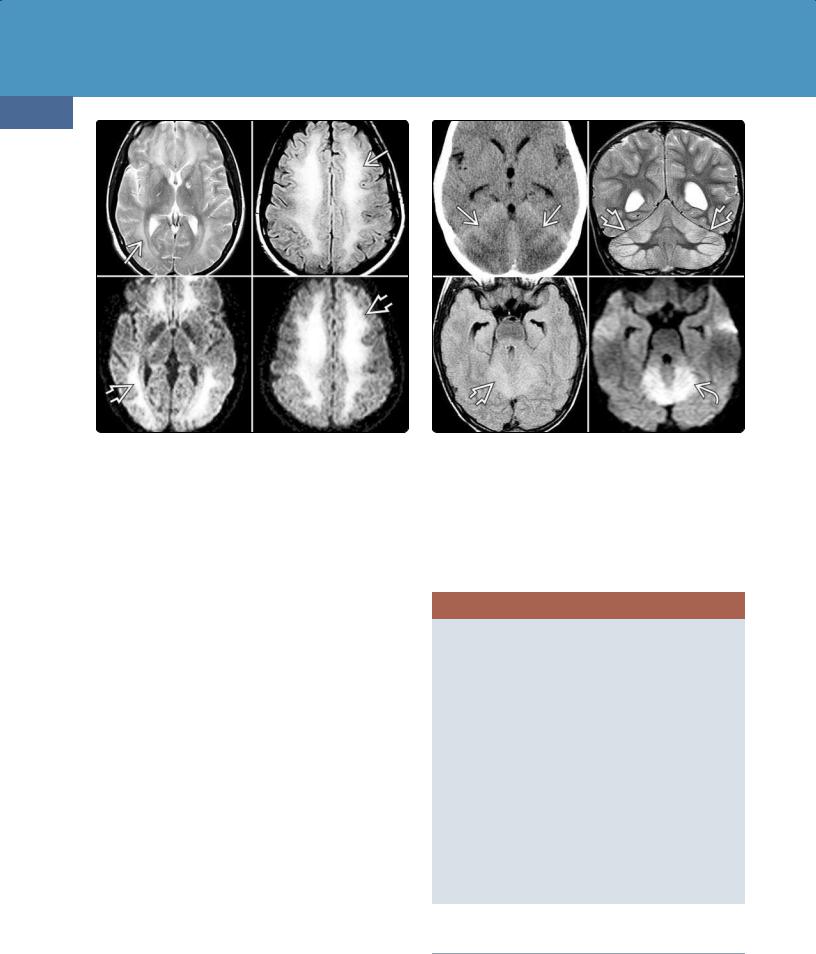
CT Findings. Acute CNS toxicity from inhaled heroin ("chasing the dragon") is characterized by symmetric hypodensities in the cerebellar white matter, sometimes described as a "butterfly wing" pattern (30-30). The posterior cerebral white matter, posterior limb of the internal capsule, and globi pallidi are also commonly affected. The anterior limb of the internal capsule is typically spared.
MR Findings. T2 and FLAIR scans in patients with early heroin-related leukoencephalopathy show symmetric hyperintensity in the cerebellar white matter with relative sparing of the dentate nuclei. There is often selective symmetric involvement of the posterior limb of the internal capsule, the corticospinal tract, the medial lemniscus, and the tractus solitarius (30-30).
Confluent hyperintensity in the cerebral white matter, including the corpus callosum, is common in severe cases of heroin vapor encephalopathy (3031). DWI shows acute diffusion restriction in the affected areas; MRS shows a lactate peak in the cerebral white matter.
Toxic Encephalopathy
935
(30-30) Inhaled heroin results in abnormalities in pons , cerebellum , corpus callosum, and internal capsules . (Courtesy K. Nelson, MD.)
(30-31) "Chasing the dragon" shows hyperintensity and restricted diffusion in periventricular WM. (Courtesy M. Michel, MD.)
(30-32) FLAIR shows symmetric cerebellar and white matter lesions in oxycodone overdose.
Toxic, Metabolic, Degenerative, and CSF Disorders
936
(30-33) MRs in a 33y woman who overdosed on methadone show striking symmetric confluent hyperintensity (leukoencephalopathy) on FLAIR and restricted diffusion on DWI .
Chronic heroin abuse can cause microvascular disease. Scattered multifocal hyperintensities in the subcortical or periventricular white matter are common but neither as prevalent nor as severe as seen with cocaine vasculopathy.
Longer duration of heroin use is also associated with more damaging effects on brain functions. fMR has demonstrated that heroin-induced brain changes may last long after abstinence.
Methadone
So-called substitute drugs such as the synthetic opioid methadone are used in the medication-assisted therapy for drug abuse/dependence as well as in the management of intractable pain. With increasing use and availability, methadone overdose is likewise growing.
A postopioid delayed toxic leukoencephalopathy similar to that caused by inhaled heroin has been reported with methadone. Diffuse, symmetric, confluent hyperintensity in the cerebral white matter on T2/FLAIR is seen (30-33). Sparing of the subcortical U-fibers is typical. In contrast to heroin toxicity, cerebellar and brainstem changes are subtle or absent in adults. MRS shows elevated choline, decreased NAA, and increased lactate.
Accidental ingestion of methadone has been reported to cause severe cerebellar edema with acute obstructive hydrocephalus in children (30-34).
(30-34) Accidental methadone poisoning in a child shows bilateral cerebellar hypodensity on NECT , T2/FLAIR hyperintensity , and restricted diffusion .
Oxycodone
Imaging in the few reported cases of oxycodone and OxyContin overdose shows restricted diffusion in the cerebellar hemispheres and globi pallidi (30-32).
OPIOID DRUGS
Heroin
•Injected
○Most common = ischemic strokes
○Globi pallidi, white matter (resembles carbon monoxide poisoning)
•Inhaled
○"Chasing the dragon"
○Most common = leukoencephalopathy
○Cerebellum, cerebral white matter
Methadone
•Adults
○Toxic leukoencephalopathy
•Children
○Usually accidental ingestion
○Cerebellar edema
Oxycodone
•Cerebellar, globus pallidus ischemia
•Less common = toxic leukoencephalopathy
Inhaled Gases and Toxins
Some drugs of abuse such as heroin have multiple potential routes of administration. Others are solely gases and therefore exclusively inhaled. Examples include toxins such as
carbon monoxide and drugs of abuse such as nitrous oxide. Some toxins such as cyanide can be inhaled, ingested, or absorbed transdermally. Cyanides may also cause—or contribute to—deaths from smoke inhalation.
Inhaled vapors from volatile, intrinsically liquid agents include amyl nitrite ("poppers") and industrial solvents (e.g., toluene). Studies have shown that petrol sniffing is often the earliest inhaled drug used and increases both the likelihood and earlier use of other drugs.
Carbon Monoxide Poisoning
Terminology
Carbon monoxide (CO) is a colorless, odorless, tasteless gas that is produced by the incomplete combustion of various fuels. CO poisoning is caused by deliberate or accidental inhalation.
Toxic Encephalopathy
937
Etiology
The toxic effects of CO result mostly from impaired oxygen transport. CO combines reversibly with hemoglobin (Hgb) with over 200 times higher the affinity than that of oxygen. If carboxyhemoglobin (CO-Hgb) levels exceed 20%, brain and cardiac damage are common.
CO-Hgb impairs erythrocyte oxygen transport, reducing cellular oxygen and causing hypoxia. In addition, lipid peroxidation leads to oxidative injury. Peroxynitrites damage the vascular endothelium.
Pathology
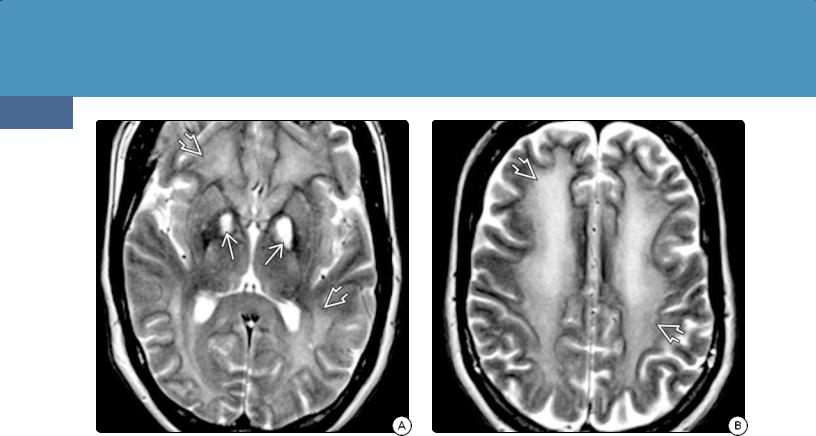
Location. Because the globi pallidi are exquisitely sensitive to hypoxia, the hallmark of acute CO poisoning is symmetric globus pallidi necrosis (30-35) (30-36). The cerebral white matter is the second most commonly affected and often
(30-35) Axial graphic shows the typical involvement of the brain by CO poisoning. The globi pallidi (GP) are most affected, followed by the cerebral white matter. Pathologically, there is necrosis of the GP with variable areas of necrosis and demyelination in the white matter. (30-36) Autopsy of carbon monoxide poisoning shows symmetric coagulative (nonhemorrhagic) necrosis of both medial GP . (Courtesy R. Hewlett, MD).
(30-37A) T1WI in a 49y man with CO poisoning shows symmetric lesions in both medial GP. Note faint hyperintense rim , thin hypointense underlying rim, and central coagulative necrosis seen as mildly hyperintense lesions . (30-37B) FLAIR scan in the same patient shows that the lesion is mostly hyperintense with central isointense core . The isointense parts of the lesions enhanced on T1 C+ (not shown).
Toxic, Metabolic, Degenerative, and CSF Disorders
938
(30-38A) Axial T2WI in a patient with CO poisoning 2 weeks prior shows characteristic bilateral hyperintensities in globi pallidi . Confluent hyperintensity now involves virtually all of the cerebral WM , except the subcortical U-fibers.
shows delayed demyelination and necrosis that may appear several weeks after the initial insult.
In addition to bilateral globi pallidi and cerebral white matter, various sites such as the cerebral cortex, cerebellum, hippocampus, amygdala, corpus callosum splenium, and insula are often involved.
Clinical Issues
Presentation and Natural History. Acute CO poisoning initially causes nausea, vomiting, headache, and impaired consciousness. Outcome depends on both duration and intensity of exposure. Seizures, coma, and death may ensue.
Patients who survive CO poisoning often develop delayed encephalopathy. Parkinson-like symptoms, memory deficits, and cognitive disturbances are common.
Treatment Options. Although there is some early success in therapies that target the downstream inflammatory and oxidative effects of CO poisoning, to date there is no available antidotal therapy. Hyperbaric oxygen significantly reduces the permanent neurologic and affective effects of CO poisoning. Early administration of 100% inspired oxygen may help mitigate long-term neuropsychiatric sequelae.
Imaging
CT Findings. Early NECT scans may be normal. Symmetric hypodensity in both globi pallidi develops within a few hours. Gross hemorrhage is rare. Variable diffuse hypodensity in the hemispheric white matter can be seen in severe cases.
MR Findings. Multiplanar MR (e.g., FLAIR, T2WI, and DWI) is the most sensitive technique for early detection of changes
(30-38B) More cephalad T2WI shows that the hyperintensity involves most of the corona radiata , mostly spares subcortical WM. This was "interval" (subacute) form of CO poisoning with toxic demyelination.
caused by CO poisoning. T1WI shows subtle hypointensity in the globi pallidi. A faint rim of hyperintensity caused by hemorrhage or coagulative necrosis may be present (30-37A).
T2/FLAIR shows bilateral hyperintensities in the medial globi pallidi (30-37B), with the putamina and caudate nuclei less commonly affected. A thin hypointense rim around the lesion may be present.
In addition to the hyperintense areas seen on T2WI, FLAIR imaging may disclose subtle involvement of the caudate nuclei, thalami, hippocampi, corpus callosum, fornices, and cerebral cortex.
DWI/ADC maps show restricted diffusion in the affected areas. Bilateral globi pallidi hyperintensities as well as foci of restricted diffusion in the subcortical white matter are typical. ADC in the cerebral white matter increases significantly, reflecting extensive microstructural tissue damage. DTI shows fractional anisotropy decline in associated cortical areas.
T2* GRE or SWI may show hypointensity in the globi pallidi suggestive of petechial hemorrhage.
Within a week after exposure, MRS shows elevated Cho/Cr and lowered NAA/Cr ratios, indicating increased membrane metabolism and decreased neuroaxonal viability.
Up to one-third of CO patients develop a delayed leukoencephalopathy with progressive white matter demyelination, the "interval" (subacute) form of CO poisoning. Extensive bilateral symmetric confluent areas of hyperintensity on T2/FLAIR are characteristic findings (30-38).

Toxic Encephalopathy
939
(30-39) Axial T2WI in a patient with nitrous oxide abuse shows selective symmetric demyelination of the posterior columns , characteristic of subacute combined degeneration. (Courtesy C. Glastonbury, MBBS.)
Differential Diagnosis
The major differential diagnoses of CO poisoning are hypoxicischemic encephalopathy (HIE) and drug abuse. As they share some common pathophysiology, imaging findings often overlap. HIE generally affects the entire basal ganglia and hippocampi, less often the white matter or only the globi pallidi. Organophosphate poisoning (accidental or suicidal exposure) can cause bilateral hemorrhagic pallidal necrosis.
Wilson disease involves the basal ganglia, mesencephalon, pons, and dentate nuclei.
Mitochondrial encephalopathies, especially Leigh disease, generally affect younger patients. Brainstem and putamen lesions are more common than globi pallidi involvement.
Some viral encephalitides, such as Japanese encephalitis, preferentially affect the basal ganglia and thalami. Creutzfeldt-Jakob disease (CJD) is rapidly progressive and affects the caudate nuclei, anterior basal ganglia, and cortex. Posteromedial thalamic involvement (pulvinar) is common in CJD and rare in CO poisoning.
Nitrous Oxide
Nitrous oxide (N O), commonly known as "laughing gas," is an inhaled anesthetic supplement commonly used in dentistry and oral surgery. N O is extremely soluble in fatty compounds and is used as an aerosol spray propellant (e.g., whipped cream canisters and cooking sprays). Anesthetic gases, including N O, are sometimes inhaled for the putative euphoria.
Excess N O irreversibly oxidizes the cobalt ion of vitamin B12, which is necessary for methylation of myelin sheath
(30-40) Sagittal T1WI shows thinned corpus callosum , and T2/FLAIR demonstrates confluent white matter hyperintensity. This was toluene toxicity due to chronic glue sniffing.
(Courtesy S. Lincoff, MD.)
phospholipids. Long-term nitrous oxide abuse causes progressive myelopathy and a peripheral polyneuropathy. The end result is subacute combined degeneration of the spinal cord. The dorsal columns and corticospinal tracts are preferentially affected (30-39). Brain lesions are rare.
Toluene Abuse
The most important component of industrial solvents is toluene, so we focus our discussion on this particular solvent. Toluene, a colorless liquid found in glues, paint thinners, inks, and other industrial products, is lipid-soluble and rapidly absorbed by the CNS. Prolonged exposure through occupation or purposeful inhalation causes multifocal neurologic defects and optic neuropathy.
Terminology
Toluene, also called methylbenzene, is an aromatic hydrocarbon. Toluene poisoning results in chronic solvent encephalopathy.
Etiology
The common methods of solvent abuse are "sniffing" (direct inhalation from a container), "huffing" (inhalation from a soaked rag held over the nose and mouth), and "bagging" (inhalation from a plastic bag).
Pathology
Toluene preferentially affects the cerebral white matter and optic nerves, causing demyelination and gliosis. Iron deposition in the thalami and basal ganglia due to demyelination and axonal loss is also common.
Toxic, Metabolic, Degenerative, and CSF Disorders
940
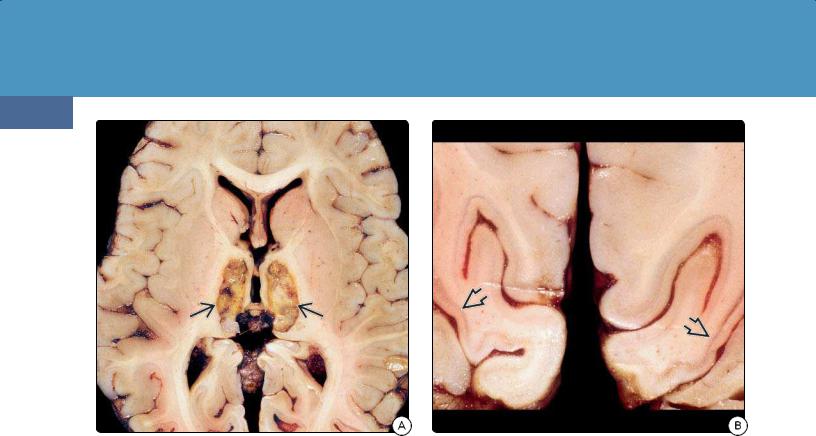
(30-41A) Autopsy specimen is from a patient with smoke inhalation, possibly from burning trash with vaporized cyanide. Note bilateral thalamic necrosis . (Courtesy R. Hewlett, MD.)
Clinical Issues
Solvent abuse is particularly prevalent among adolescents and young adults. Low cost and ease of access have led to increased prevalence in many countries. Regular long-term toluene abuse causes severe and irreversible cognitive impairment.
Imaging
Imaging in patients with acute toluene abuse is usually normal. Abnormalities are typically seen only after several years of chronic inhalant abuse. Diffuse white matter lesions are seen in nearly half of all patients, initially seen as T2/FLAIR hyperintensity in the deep periventricular white matter with subsequent spread into the centrum semiovale and subcortical areas. The internal capsule, cerebellum, and pons are often affected (30-40).
Chronic prolonged toluene exposure also causes generalized atrophy with ventricular dilatation and enlarged subarachnoid spaces. White matter volume loss is seen as thinning of the corpus callosum. The extent of volume loss directly correlates with abuse duration.
Organophosphate Poisoning
Organophosphates (OPs) are common ingredients in pesticides. Because of their widespread agricultural use, ready availability, and easy accessibility, OPs are potential sources of accidental or suicidal exposure.
"Street pesticides" (illegal, unlabeled, and decanted agricultural pesticides used predominantly for urban
(30-41B) Coronal section through the occipital lobes of the same patient shows cortical laminar necrosis . (Courtesy R. Hewlett, MD.)
household purposes) pose an increasing risk for significant pesticide exposures and poisonings in emerging nations.
The anticholinesterase effect of OPs causes three potential discrete neurologic syndromes. The initial acute effect is a lifethreatening acute cholinergic crisis due to excessive stimulation of muscarinic receptors. The intermediate syndrome is characterized by cranial nerve palsies, proximal muscle weakness, delayed polyneuropathy, and Parkinson-like extrapyramidal symptoms.
Chronic or low-dose occupational exposure may result in neurobehavioral and neuropsychiatric disorders. There is increasing evidence that some cases of acquired amyotrophic lateral sclerosis (ALS) are linked to OP exposure.
Acute OP poisoning causes hemorrhagic basal ganglia necrosis with the "eye of the tiger" sign. On T2WI, a ring of marked hypointensity caused by excess iron accumulation surrounds a central hyperintense focus in the medial globus pallidus.
The differential diagnosis of OP poisoning includes other drug-induced causes of pallidal necrosis such as carbon monoxide poisoning. HIE and metabolic encephalopathies such as Leigh and Wilson diseases also affect the basal ganglia.
Cyanide Poisoning
Terminology
Cyanide (CN) is one of the most potent and deadly of all poisons. Cyanogenic compounds may be found in household or workplace substances and deliberately or accidentally ingested.

Toxic Encephalopathy
941
(30-42A) FLAIR scan in smoke inhalation with CN poisoning from burning plastic shows symmetric hyperintensity in caudate nuclei and putamina , more subtle lesions in posteromedial thalami , and curvilinear cortical hyperintensities .
Acute CN intoxication is also called CN poisoning and is often the result of attempted suicide or smoke inhalation. Chronic CN toxicity is usually caused by occupational exposure to substances that contain cyanogenic compounds. Chronic CN exposure results in cyanide encephalopathy.
Etiology
CN exists in gas, solid, and liquid form. CN poisoning can occur by inhalation, ingestion, or transdermal absorption. Combustion of many common materials such as some fabrics and plastics may release CN and cyanogenic compounds. Cyanogenic compounds are also found in some foods, including almonds, the pits of stone fruits, Lima beans, and cassava root.
CN inactivates cytochrome oxidase, a key enzyme in the mitochondrial respiratory chain. Therefore, acute CN poisoning typically affects structures with high metabolic requirements. The basal ganglia and cortex are most commonly involved. Cerebral hypoxia may occur as part of the acute intoxication process, complicating both the diagnosis and treatment of CN poisoning.
Pathology
Hemorrhagic basal ganglia necrosis and laminar cortical necrosis are the pathologic hallmarks of CN poisoning (3041).
Clinical Issues
(30-42B) More cephalad scan in the same patient shows the cortical hyperintensities , which are especially prominent in both occipital lobes .
CN poisoning is fatal in 95% of cases with death often occurring in minutes. Survivors may develop pseudoparkinsonism with extrapyramidal symptoms.
INHALED GASES AND TOXINS
Carbon Monoxide Poisoning
•Acute: symmetric globi pallidi necrosis
•Subacute ("interval"): confluent leukoencephalopathy
Nitrous Oxide Abuse
•Brain lesions rare
•Subacute combined degeneration of the spinal cord
○Hyperintensity in dorsal columns
Toluene (Solvent) Abuse
•Chronic, repeated use
○Atrophy
○White matter lesions
○Thalami, substantia nigra, red nuclei, dentate lesions
Organophosphate (Pesticide) Poisoning
•Basal ganglia hemorrhage, necrosis
•"Eye of the tiger" sign
Cyanide Poisoning
•Suicide, smoke inhalation
•Basal ganglia hemorrhage, necrosis
•Laminar cortical necrosis
Imaging
Patients with acute CN poisoning typically present with unresponsiveness, hemodynamic instability, and severe lactic acidosis. Because administered doses are usually high, acute
MR is the modality of choice to depict lesion extent. Patients who survive the initial insult show symmetric hyperintensity in the basal ganglia and linear cortical hyperintensity on T2WI

Toxic, Metabolic, Degenerative, and CSF Disorders
942
(30-43) NECT scan demonstrates volume loss in the frontal and temporal lobes attributable to lead poisoning. (Courtesy R. Ramakantan, MD.)
(30-44) Autopsy case of chronic mercury poisoning shows diffuse cortical, cerebellar volume loss. The medulla, pons, and midbrain also appear shrunken. (Courtesy R. Hewlett, MD.)
and FLAIR (30-42). CN poisoning usually spares the hippocampi. T1 C+ scans typically show intense enhancement in the affected areas.
In the subacute and chronic stages, hemorrhagic necrosis causes T1 hyperintensity in the basal ganglia. Laminar necrosis results in serpentine linear hyperintensity in the cortex.
Differential Diagnosis
The most important differential diagnosis of CN poisoning is hypoxic-ischemic encephalopathy. It may complicate CN poisoning, and their features often overlap because the basal ganglia are affected in both disorders.
Metal Poisoning and
Toxicity
A variety of metals can cause serious neurologic dysfunction when deposited in excess amounts in the CNS. Manganese accumulation is more common in the setting of chronic liver failure (see Chapter 32) but also occurs with occupational exposure. Other environmental toxins such as lead and mercury can cause significant neurotoxicity.
Lead Poisoning
Lead (Pb) is a potent and pervasive environmental neurotoxicant that is especially harmful during childhood development. Chronic Pb poisoning occurs in three forms: (1) a gastrointestinal form (anorexia, vomiting, lead "colic," etc.),
(2) a neuromuscular form (muscle weakness, myalgias, peripheral neuritis, etc.), and (3) a cerebral or
neuropsychiatric form (irritability, headache, encephalopathy, seizure, etc.). The cerebral form is common in children, whereas neuromuscular manifestations are more common in adults. The gastrointestinal form occurs in both age groups.
Lead-containing cooking utensils and indigenous medications are common sources of Pb poisoning in developing nations. Chronic lead exposure is associated with a significant and persistent impact on white matter microstructure.
Patients with moderate to severe lead encephalopathy usually have blood lead levels that exceed 70 μg/dL. In such cases, CT or MR may reveal volume loss, especially in the frontal cortex and subcortical WM (30-43).
In less severe cases, DTI may reveal subtle changes such as decreased fractional anisotropy and diffusivity in the corona radiata and corpus callosum.
Mercury Poisoning
Mercury (Hg) occurs naturally in three forms: elemental Hg, mercury vapor, and organic/inorganic. Elemental mercury ("quicksilver") is liquid at room temperature. Liquid Hg is not absorbed through the skin and, if swallowed, passes through the GI tract without being absorbed. Mercury vaporizes easily, is highly diffusible, lipid soluble. Hg vapor is very toxic and easily absorbed.
Although occupational exposures to Hg still occasionally occur in manufacturing and mining, most current cases are caused by dermal absorption from illegal skin-lightening cosmetic products or bioconcentration of inorganic methylmercury in the food chain. Seafood (fish, marine mammals) is especially

susceptible to contamination. Organic mercury poisoning is known as Minamata disease.
Gross pathology shows widespread cortical atrophy, white matter shrinkage, and thinning of the corpus callosum (3044). Severe spongiosis and gliosis with neuronal loss are seen on microscopic examination.
Imaging findings of Minamata disease include atrophy of the calcarine (visual) cortex, cerebellar vermis and hemispheres, and the postcentral cortex. Decreased regional blood flow in the cerebellum can be demonstrated even in the absence of cerebellar atrophy.
Treatment-Related
Disorders
A comprehensive treatment of all iatrogenic abnormalities in the brain is far beyond the scope of this text. Here we discuss the most common disorders with a focus on treatment effects that must be recognized on imaging studies, namely radiation, chemotherapy, and surgery.
Radiation Injury
In the United States, approximately 100,000 primary and metastatic brain tumor patients each year survive long enough (more than 6 months) to develop some degree of radiation-induced injury (RII) to the brain.
CNS response to stressors and injuries such as ionizing radiation are modulated by responses of the brain's innate immune effector cells, the microglia. Exposure to high doses of ionizing radiation leads to the expression and release of proinflammatory cytokines and reactive oxygen species, leading to the tissue destruction that occurs with RII.
Many investigators divide RII into three phases: acute injury, early delayed injury, and late delayed injury. However, the pathophysiology and natural course of radiation therapy (XRT)-induced CNS injury are not well understood. Pathologically, radiation injury varies from mild transient vasogenic edema to frank necrosis. The damage that results from XRT depends on a number of variables including total dose, field size, number/frequency/fractionation of doses, whether chemotherapy is used in conjunction with XRT, and patient age.
Several different CNS tissues are affected by XRT. Vascular endothelial cells, oligodendrocytes, astrocytes, microglia, and neurons probably all interact in the brain's response to radiation injury.
Oligodendrocytes are especially sensitive. Vascular injury occurs in both early and late delayed injury. Once considered relatively radioresistant, neurons are now known to respond negatively to radiation and probably play a significant but as- yet-unidentified role in late radiation-induced cognitive impairment.
Toxic Encephalopathy
943
Acute Radiation Injury
Acute RII occurs days to weeks after irradiation and is very rarely encountered with modern XRT regimens. The major clinical manifestations of acute RII include headache and drowsiness.
Standard imaging studies are usually normal, although MRS, DTI, and fMR may detect changes before neurocognitive symptoms or anatomic alterations emerge. Occasionally, transient white matter edema secondary to changes in capillary permeability can be seen on T2/FLAIR sequences.
Biomarkers of pathology such as the mitochondrial translocator protein 18 kDa (TSPO) have facilitated in vivo characterization of microglial activation. In the future, microglial PET imaging may be a potential biomarker of early neuroinflammation from radiation-induced brain injury.
Early Delayed Radiation Injury
In early delayed RII, imaging abnormalities can be detected as early as 1 to 6 months after XRT is completed. Early delayed RII is characterized pathologically by transient demyelination and clinically by somnolence, attention deficits, and shortterm memory loss. Patients may have significant cognitive impairments even in the absence of detectable anatomic abnormalities.
Confluent hypodense areas on NECT and periventricular white matter hyperintensity on T2/FLAIR are typical abnormalities. At this stage, RII changes are generally mild and reversible, often resolving spontaneously.
Late Delayed Radiation Injury
Late delayed RII is usually not observed until at least 6 months post irradiation. These late delayed injuries are viewed as progressive and largely irreversible, resulting from loss of glial and vascular endothelial cells.
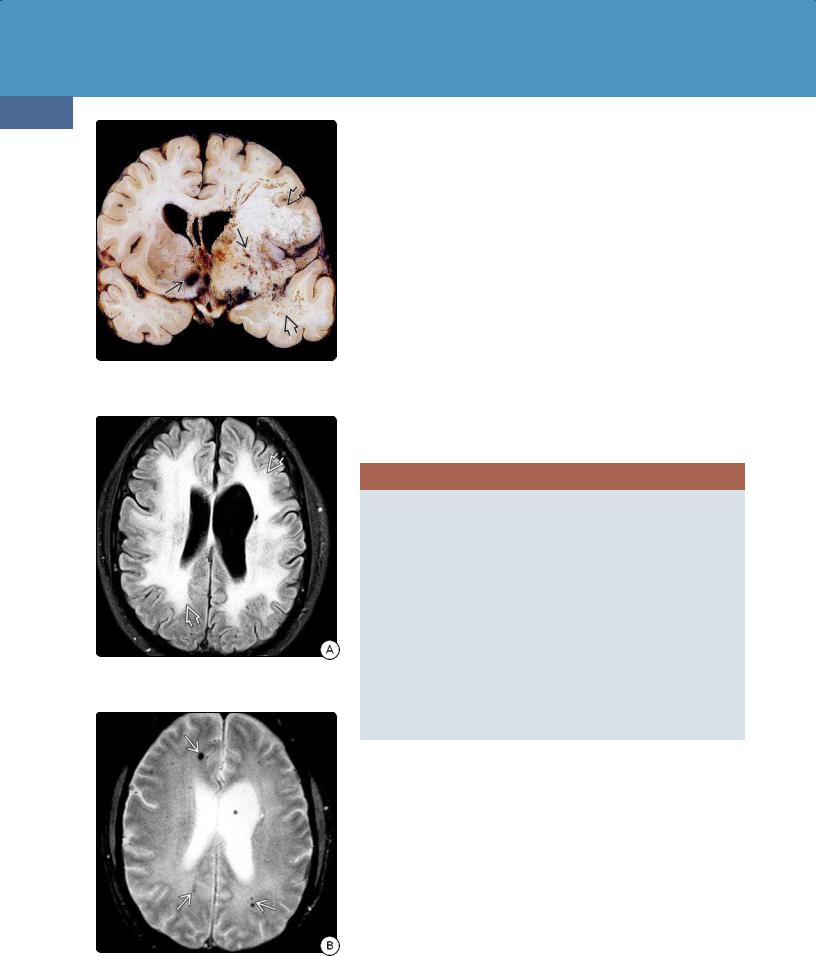
Pathologically, coagulative necrosis in a "mosaic" pattern with coalescing foci produces a necrotizing leukoencephalopathy in the deep cerebral white matter. The subcortical association or U-fibers and corpus callosum are typically spared (30-45).
Vascular changes include fibrinoid necrosis, hyalinization, and sclerosis with thrombosis. Late delayed radiation necrosis is initially expansile and mass-like, with necrosis largely confined to white matter.
Initially, late delayed RII shows mass effect and variable enhancement on imaging studies. Later, volume loss, white matter spongiosis with confluent hyperintensity, and calcifications can be seen (30-46).
Long-Term Sequelae of Radiation Injury
In addition to necrotizing leukoencephalopathy, long-term complications of XRT include vasculopathy, mineralizing microangiopathy, microvascular glomeruloid proliferation with telangiectasis (XRT-induced vascular malformations), and the development of radiation-induced neoplasms.
Toxic, Metabolic, Degenerative, and CSF Disorders
944
(30-45) Autopsy shows XRT-induced necrotizing leukoencephalopathy , hemorrhagic vascular malformations . (Courtesy R. Hewlett, MD.)
(30-46A) FLAIR in a patient with cognitive decline 3 years after whole-brain XRT for leukemia shows confluent WM hyperintensity .
(30-46B) T2* scan shows multiple foci of gradient blooming , necrotizing leukoencephalopathy with XRT-induced vascular malformations.
Radiation-induced vasculopathy with endothelial hyperplasia results in diffusely narrowed large and medium-sized arteries. Ischemic strokes and moyamoya-like disease may result (30-47).
Mineralizing microangiopathy is usually seen in patients treated with combination XRT and chemotherapy. Mineralizing microangiopathy generally does not appear until at least 2 years following treatment; it is then seen as calcifications in the basal ganglia and subcortical white matter (3048).
Radiation-induced vascular malformations (RIVMs) are primarily capillary telangiectasias or cavernous malformations, most commonly seen in children who have received whole-brain radiotherapy for acute lymphoblastic leukemia. T2* (GRE, SWI) sequences demonstrate "blooming" microhemorrhages in the majority of patients (30-49). It is uncommon to develop RIVMs less than 3 years following XRT. Children under 10 years of age at the time of irradiation are at higher risk.
Radiation-induced neoplasms are rare but often devastating. XRT is the single most important risk factor for developing a new primary CNS neoplasm. Approximately 70% are meningiomas, 20% malignant astrocytomas or medulloblastomas, and 10% sarcomas. Meningiomas occur an average of 17-20 years after treatment, whereas gliomas occur at a mean of 9 years. Sarcomas have a mean latency of 7 or 8 years following XRT.
RADIATION-INDUCED BRAIN INJURY
Pathology
•Microglial activation
•Proinflammatory cytokines
Three Phases of RII
•Acute radiation injury
○Rare
○T2/FLAIR may show white matter edema
○TSPO-PET may show neuroinflammation
•Early delayed injury (at least 6 months)
○Necrotizing leukoencephalopathy
○Confluent hyperintensity
•Long-term sequelae
○Necrotizing leukoencephalopathy
○Vasculopathy, mineralizing microangiopathy
○Vascular malformations (T2* "black dots")
○Radiation-induced neoplasms
Chemotherapy Effects
Currently, the most common chemotherapy agents implicated in CNS toxicity are methotrexate, cytarabine, vincristine, asparaginase, and corticosteroids.
Unlike radiation injury, chemotherapy-associated acute toxic CNS injury is common. The two most frequent abnormalities are posterior reversible encephalopathy syndrome and treatment-induced leukoencephalopathy.
Posterior reversible encephalopathy syndrome (PRES) is addressed in detail in Chapter 32. In chemotherapy-related PRES, imaging findings are often atypical. The occipital lobes are frequently spared whereas the cerebellum, brainstem, and basal ganglia are frequently involved. Hemorrhage, contrast enhancement, and diffusion restriction—all relatively rare in "typical" PRES—are common.

Toxic Encephalopathy
945
(30-47) MRA in a case with right MCA stroke years after XRT shows moyamoya and postradiation vasculopathy. Stenosis of both supraclinoid ICAs is present; right MCA is occluded. (Courtesy P. Hildenbrand, MD.) (30-48) NECT shows 20y man with XRT and chemo at age 8 for medulloblastoma. BG, subcortical WM calcifications are characteristic of mineralizing microangiopathy. (Courtesy P. Chapman, MD.)
(30-49A) Patient with whole-brain radiation, chemo for anaplastic oligodendroglioma presented with seizure 5 years after treatment. T1 C+ FS shows multiple enhancing foci in left hemispheric WM ; delayed radiation necrosis vs. tumor recurrence. (3049B) pMR shows elevated rCBV in the enhancing areas , suggesting the enhancing foci represent recurrent tumor rather than necrotizing leukoencephalopathy.
(30-49C) T2* SWI scan shows innumerable "blooming" hypointense foci in the WM, consistent with radiation-induced vascular malformations. (30-49D) More cephalad T2* SWI scan in the same patient shows additional small lesions and a larger focus , consistent with hemorrhage into the recurrent neoplasm. Biopsy confirmed recurrence of anaplastic oligodendroglioma (WHO grade III), capillary telangiectases.
Toxic, Metabolic, Degenerative, and CSF Disorders
946
CHEMOTHERAPY EFFECTS ON THE BRAIN
Clinical Issues
•Acute effects common
•Often reversible
Imaging
•PRES common
○Atypical > typical imaging findings
○Occipital lobes often spared
○Hemorrhage, enhancement, restricted diffusion common
•Acute leukoencephalopathy
○Reflects acute neurotoxicity
○Transient T2/FLAIR periventricular hyperintensity
lymphoblastic leukemia. Bilateral, relatively symmetric, confluent areas of T2/FLAIR hyperintensity in the periventricular white matter are typical (30-50). Imaging abnormalities typically resolve after treatment.
Effects of Surgery
Interpreting imaging findings in the postoperative brain can be challenging. Expected findings include pneumocephalus, focal hemorrhage, retraction edema, small subdural CSF collections (hygromas), etc. We focus on just two abnormalities that are important to recognize on imaging studies: retained surgical material ("textiloma") and sinking skin flap syndrome.
Textiloma
Treatment-induced leukoencephalopathy is especially common in patients treated with methotrexate. Acute neurotoxicity occurs in 5-18% of children treated for acute
(30-50A) T2WI in a 5y child with acute deterioration following intrathecal methotrexate for ALL shows symmetric confluent hyperintensity in the deep periventricular WM . Note sparing of subcortical U-fibers. (3050B) More cephalad scan in the same patient shows the confluent WM hyperintensity and spared subcortical WM. This is methotrexateinduced leukoencephalopathy.
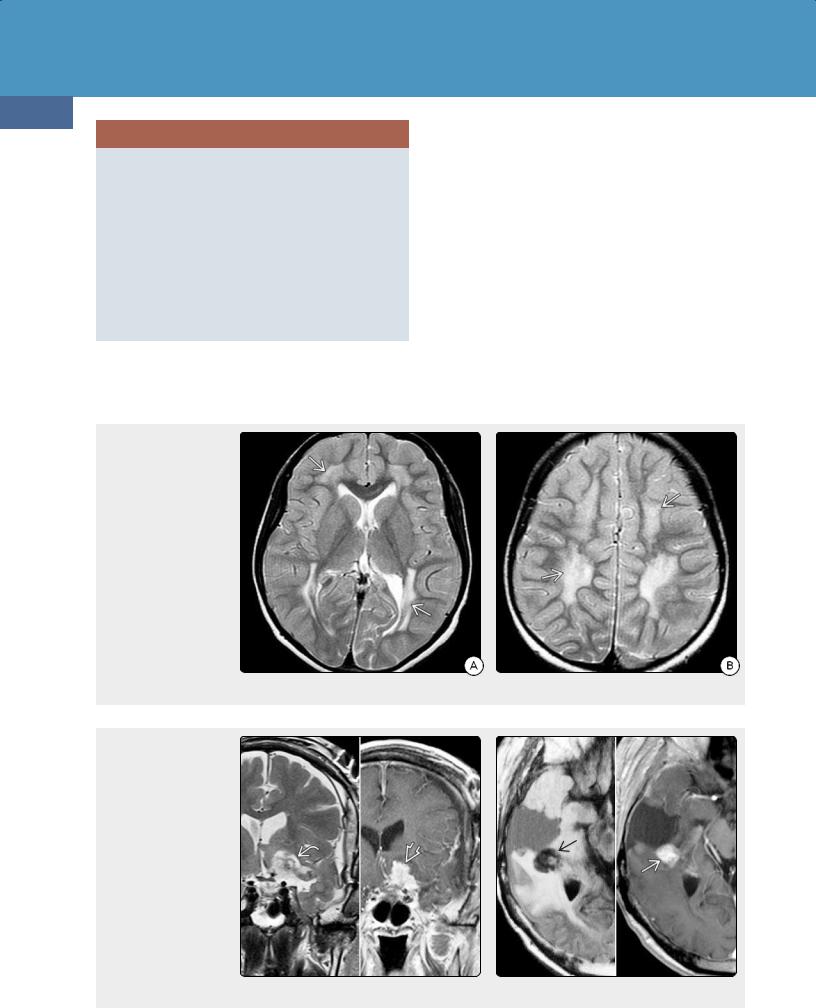
Textiloma—also known as muslinoma or gauzoma—is a foreign body reaction to retained surgical elements. The term has traditionally referred to reactions to surgical elements
(30-51) T2WI (L), T1 C+ (R) after meningioma resection show that a mixed signal mass enhances strongly and uniformly ; this is textiloma. (30-52) (L) Retained cotton ball after surgery is round and hypointense on FLAIR .
(R) T1 C+ shows that the retained material enhances strongly but heterogeneously . This is textiloma. (Courtesy B. K. KleinschmidtDeMasters, MD.)
inadvertently left in the operative bed but has recently been expanded to include reactions to intentionally placed surgical elements.
Intracranial textilomas are rare. Both resorbable and nonresorbable hemostatic agents may be placed in the surgical bed to provide persistent hemostasis after closure.
When they occur, textilomas can be mistaken for recurrent tumor or abscesses on imaging studies. Most surgically placed materials [e.g., cotton hemostats, muslin, or polytetrafluoroethylene (Teflon)] do not have signal abnormalities on MR and are visualized only when a foreign body reaction develops, forming a textiloma.
Nearly 40% of textilomas are hypointense on T2WI and FLAIR (30-51). Many "bloom" on T2* (GRE, SWI) sequences. Restriction on DWI is variable. Most enhance; both solid and ring patterns occur (30-52).
Sinking Skin Flap Syndrome
Sinking skin flap syndrome (SSFS)—also referred to as "syndrome of the trephined"—is an unusual cause of neurologic deterioration in patients who have undergone large decompressive craniectomy for uncontrollable brain swelling (usually following trauma or "malignant" hemispheric infarction).
The mechanism of SSFS has not been fully elucidated. Some authors suggest that the large cranial defect allows external (atmospheric) pressure to act on the brain, causing altered CSF dynamics. Significantly smaller CSF volumes have been measured in patients with SSFS. CSF leakage or ventriculoperitoneal shunt overdrainage can exacerbate intracranial hypovolemia and result in clinical deterioration.
SSFS occurs in 20-25% of patients who survive decompressive surgery and in whom restorative cranioplasty is delayed. It typically presents weeks to months after craniectomy but most commonly occurs during the second postoperative month.
Presenting signs and symptoms vary, but the overwhelming majority of patients exhibit a visibly sunken skin flap. In severe cases, decreased consciousness and hemiparesis can occur. SSFS can become life-threatening, especially if exacerbated by CSF leak. Symptoms typically improve after cranioplasty.
Imaging shows skin flap depression below the level of the calvarium, often with an S-shaped configuration. Mass effect on the cortex, evidenced by sulcal effacement and buckling of the gray-white matter interface under the skin flap, is seen in nearly all cases.
Paradoxical deviation of midline structures away from the craniectomy site is typical. Midline shift of the interhemispheric fissure and/or septi pellucidi away from the sunken skin flap is seen in 75% of cases (30-53).
Toxic Encephalopathy
947
(30-53A) SSFS is 5 weeks after decompressive craniectomy. Duraplasty is sunken with S-shaped configuration , mass effect on ventricles .
(30-53B) Bone CT shows the craniectomy defect and S-shape configuration of the sunken skin flap.
(30-53C) Bone CT with 3D SSD shows the extent to which the brain and duraplasty have sunken inward below the level of the craniectomy defect.