

- •Pineal Parenchymal Tumors
- •Germ Cell Tumors
- •Selected References
- •Medulloblastoma
- •Selected References
- •Anatomy of the Cranial Meninges
- •Meningomas
- •Primary Melanocytic Lesions
- •Other Related Neoplasms
- •Selected References
- •Cranial Nerve Anatomy
- •Schwannomas
- •Neurofibromas
- •Selected References
- •Histiocytic Tumors
- •Selected References
- •Sellar Region Anatomy
- •Normal Imaging Variants
- •Congenital Lesions
- •Neoplasms
- •Miscellaneous Lesions
- •Selected References
- •Intracranial Pseudotumors
- •Selected References
- •Metastatic Lesions
- •Paraneoplastic Syndromes
- •Selected References
- •Scalp Cysts
- •Extraaxial Cysts
- •Parenchymal Cysts
- •Intraventricular Cysts
- •Selected References
- •Anatomy and Physiology of the Basal Ganglia and Thalami
- •Selected References
- •Alcohol and Related Disorders
- •Opioids and Derivatives
- •Inhaled Gases and Toxins
- •Selected References
- •Selected References
- •Hypertensive Encephalopathies
- •Glucose Disorders
- •Thyroid Disorders
- •Seizures and Related Disorders
- •Miscellaneous Disorders
- •Selected References
- •The Normal Aging Brain
- •Dementias
- •Degenerative Disorders
- •Selected References
- •Normal Variants
- •Hydrocephalus
- •CSF Leaks and Sequelae
- •Selected References
- •Cerebral Hemisphere Formation
- •Imaging Approach to Brain Malformations
- •Posterior Fossa Anatomy
- •Chiari Malformations
- •Hindbrain Malformations
- •Selected References
- •Commissural Anomalies
- •Malformations Secondary to Abnormal Postmigrational Development
- •Selected References
- •Anencephaly
- •Holoprosencephaly
- •Holoprosencephaly Variants
- •Related Midline Disorders
- •Holoprosencephaly Mimics
- •Selected References
- •Selected References
- •Selected References
- •Cephaloceles
- •Craniosynostoses
- •Meningeal Anomalies
- •Selected References
- •Index

Congenital Malformations of the Skull and Brain
1174
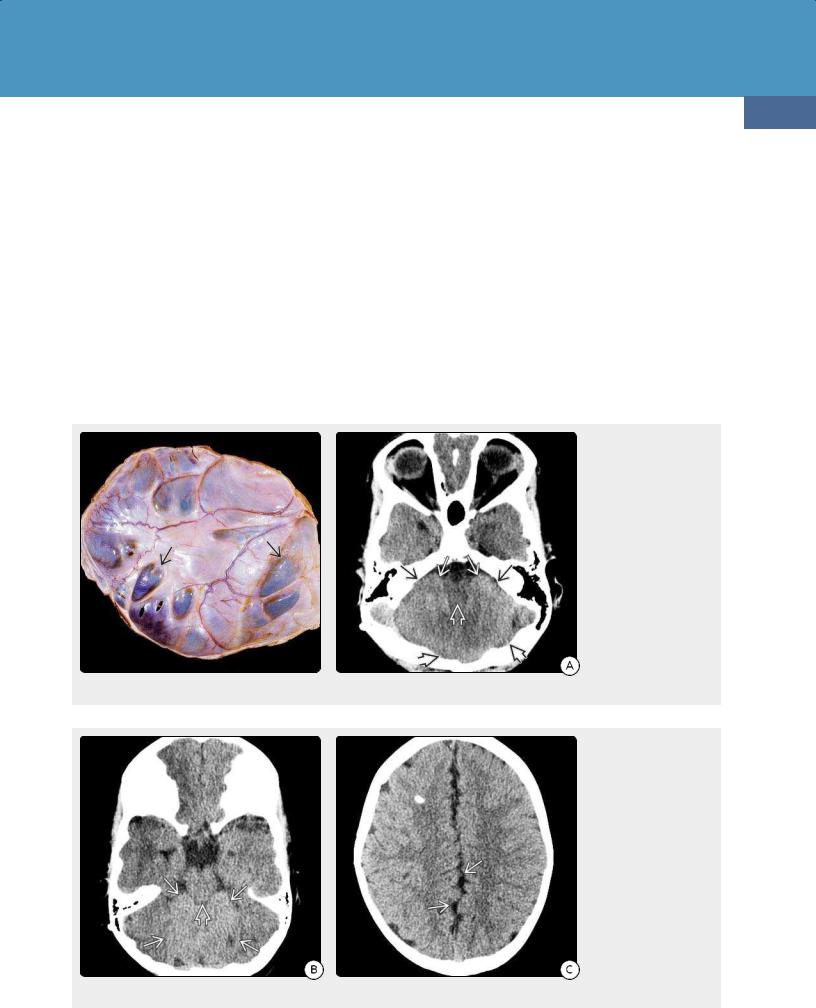
(36-13) Chiari 1 malformation shows the basion-opisthion line shown in green. Note the low-lying, pointed tonsil with vertically oriented folia . The nucleus gracilis is inferiorly displaced.
(36-14) Semiaxial view of autopsy case shows Chiari 1 malformation. Note inferiorly displaced tonsils with vertically oriented folia . (Courtesy E. T. Hedley-Whyte, MD.)
Coronal Plane
Images through the anterior "belly" of the pons show the large biventral lobules of the inferior cerebellar hemispheres with the cerebellar tonsils projecting inferomedially (36-9) (36-10). The flocculi lie just in front of the horizontal fissure
(36-9).
Moving posteriorly, the rhomboid or diamond shape of the fourth ventricle can be appreciated. Thin caps of CSF, the posterior superior recesses, cover the tops of the cerebellar tonsils (36-11). Inferiorly, the fourth ventricle opens into the cisterna magna via the foramen of Magendie. The large middle cerebellar peduncles are seen along the sides of the fourth ventricle. More posteriorly, the vermis can be seen lying between the two hemispheres (36-12).
Chiari Malformations
Introduction to Chiari Malformations
Chiari malformations were first described in the late nineteenth century by the Austrian pathologist Hans Chiari. He described what seemed to be a related group of hindbrain malformations associated with hydrocephalus and divided them into three types: Chiari 1-3.
Chiari 1 and 2 are pathogenetically distinct disorders. Chiari 1 involves inferior dislocation of the cerebellar tonsils (36-13); Chiari 2 is always associated with myelodysplasia and involves herniation of the medulla and vermis. Chiari 3 is classically characterized as herniation of posterior fossa contents through a low occipitocervical bony defect.
"Chiari 4 malformation" was originally used to designate what is now recognized as primary cerebellar agenesis, not a hindbrain herniation. The term has been abandoned.
Some authors have expanded the Chiari spectrum to include variants such as Chiari 0 (syrinx without frank tonsillar herniation), Chiari 1.5 (caudal protrusion of brainstem in addition to the tonsils), and Chiari 5 (Chiari 2 plus occipital or high cervical myelomeningocele). These variants are controversial and are briefly discussed at the end of this section.
Chiari 1
Overview
Type 1 Chiari malformation (CM1) is defined as caudal cerebellar tonsillar ectopia. However, the precise distance of the tonsils below the foramen magnum (FM) required to diagnose CM1 is not agreed upon. Some investigators consider tonsillar ectopia measuring 5 mm or more as sufficient to establish the diagnosis of CM1. However, other investigators insist additional abnormalities such as tonsillar deformity, obliterated retrotonsillar CSF spaces, or altered CSF flow dynamics should also be present.
Abnormalities of the cervical spinal cord are common in CM1. A complex CSF-filled cavity with multiple septations of spongy glial tissue is typical. Glia-lined cord cavitations are generally referred to as syringomyelia. The term hydromyelia refers to an ependyma-lined expansion of the central canal. In CM1, extensive areas of ependymal denuding and astrocytic scarring make it difficult to distinguish between hydroand syringomyelia even on histologic examination. Therefore, these nonneoplastic, septated, paracentral, fluid-containing
Posterior Fossa Malformations
1175
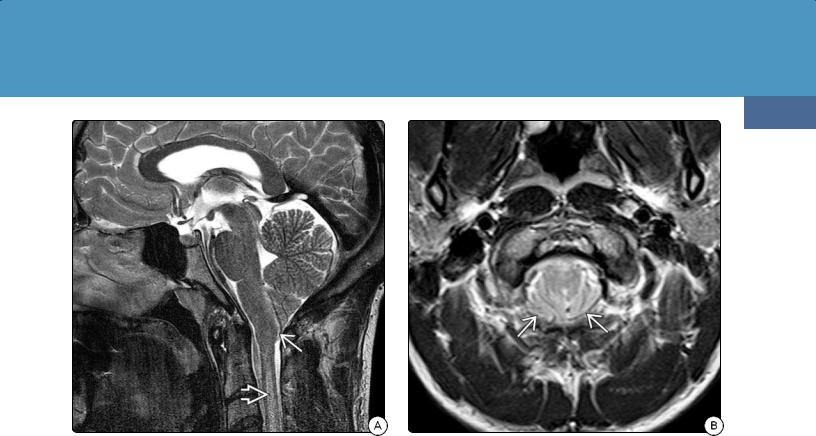
(36-15A) Sagittal T2WI in a 23y man with classic Chiari 1 malformation shows a low-lying, pointed tonsil and normalsized posterior fossa. Cord T2 hyperintensity represents "presyrinx" state.
cavitations are often referred to as hydrosyringomyelia or simply syrinx.
Etiology
General Concepts. The pathogenesis of CM1 is incompletely understood and remains controversial. Genetic, nongenetic, and epigenetic factors have all been proposed.
Primary paraxial mesodermal insufficiency with underdeveloped occipital somites has also been invoked to explain the development of CM1. Other theories suggest that disorders of neural crest-derived elements could lead to hyperor hypoossification of the basi-chondro-cranium, resulting in morphometric changes in the posterior fossa.
A combination of altered bony anatomy and abnormal CSF hydrodynamics is the most widely accepted concept.
Abnormal Posterior Fossa. Many—but by no means all—patients with CM1 demonstrate abnormal geometry of the bony posterior fossa ("normal-sized hindbrain housed in a too-small bony envelope"). Various combinations of congenitally reduced clival length, shortened basiocciput, and craniovertebral junction (CVJ) fusion anomalies may all result in diminished posterior cranial fossa depth and/or an abnormally small posterior fossa volume.
Altered CSF Dynamics. Syringomyelia is present in 40-80% of individuals with symptomatic CM1. Its etiology is also a controversial subject. Tonsillar impaction and posterior arachnoid adhesions cause increased resistance to CSF flow between the intracranial and spinal subarachnoid spaces. Systolic piston-like descent of the impacted tonsils may create abnormal intraspinal CSF pressure waves, which in turn could
(36-15B) Axial T2WI in the same patient shows "crowded" foramen magnum with obliterated retrotonsillar CSF spaces .
result in the development of hydrosyringomyelia in the upper cervical cord.
Altered CSF hydrodynamics with accelerated flow velocity and increased pressure gradients may also cause or contribute to the displacement of brain tissue out of the cranium and into the upper cervical canal.
Genetics. Familial aggregation studies, twin studies, the cosegregation of CM1 with known genetic conditions such as achondroplasia and Klippel-Feil syndrome, and recent genome-wide analyses all provide strong evidence for a genetic component of CM1.
Although the specific genetic causes of CM1 are not yet fully elucidated, chromosomes 9q21 and 15q21 (also the site of the fibrillin-1 gene, the major cause of Marfan syndrome) have been implicated in some studies. Researchers have identified five regions of the X chromosome linked to FG syndrome, which is also associated with increased prevalence of CM1.
Pathology
Grossly, the herniated tonsils in CM1 are inferiorly displaced and grooved by impaction against the opisthion (36-14). They often appear firm and sclerotic. Arachnoid thickening and adhesions around the CVJ are common. Microscopically, degenerative changes with Purkinje and granular cell loss may be present.
Clinical Issues
Epidemiology and Demographics. CM1 is the most common of the Chiari malformations and can be identified in patients of all ages. Its estimated prevalence in the general population is 0.6-1.0%, but recent studies have found a Chiari 1
Congenital Malformations of the Skull and Brain
1176
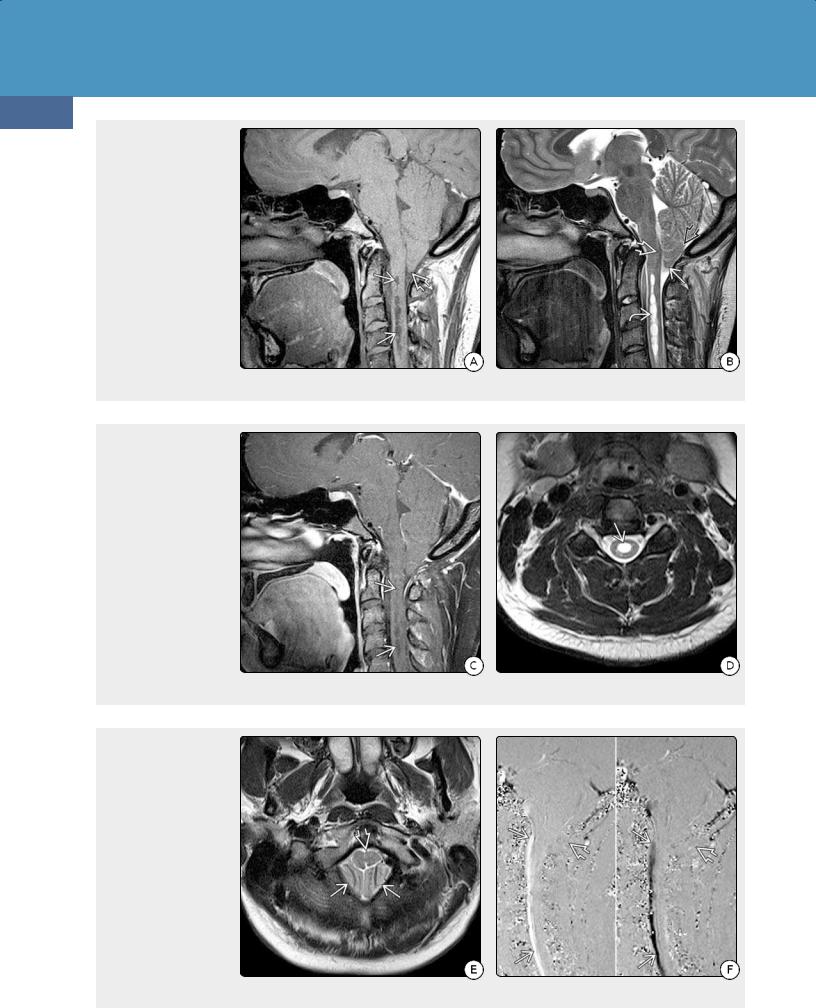
(36-16A) Sagittal T1WI shows "crowded" foramen magnum and tonsillar ectopia with the "pointed" appearance that is typical of Chiari 1. Note syrinx in the upper cervical cord . (36-16B) Sagittal T2WI shows "pointed" tonsil and obliquely oriented tonsillar folia . Multiple septations in the syrinx cavity are clearly seen . The inferior fourth ventricle is somewhat elongated, and the nucleus gracilis is slightly low-lying.
(36-16C) Sagittal T1 C+ FS shows that the syrinx does not enhance. (3616D) Axial T2WI shows a well-demarcated CSF cavity in the middle of the central cervical spinal cord. It is not possible by imaging to distinguish between ependyma-lined dilatation of the central canal (hydromyelia) and glia-lined paracentral cord cavitations (syringomyelia), so the term hydrosyringomyelia is generally used to designate this CM1associated finding.
(36-16E) T2WI in the same patient shows tonsils and a compressed and slightly deformed medulla, giving the appearance of the "crowded" foramen magnum typical of CM1. (36-16F) Sagittal phasecontrast CSF flow study in systole (L) and diastole (R) shows normal CSF flow in front of the cervicomedullary junction and no posterior flow in the foramen magnum . Tonsillar "crowding" and adhesions prevent normal CSF circulation.
Posterior Fossa Malformations
1177
(36-17) Sagittal T1WI shows severe Chiari I malformation. The tonsils are pointed inferiorly , lying 15 mm below the crowded foramen magnum. The obex and nucleus gracilis are kinked and displaced inferiorly . The odontoid is retroflexed .
malformation in 3.6% of children undergoing routine brain or cervical spine MR. There is no sex predilection.
A CM1-associated syrinx is rare in infants under 1 year, but the incidence rises with age. This age-related increase is most pronounced in the first 5 years of life.
Presentation. One-third to half of all patients with CM1 on imaging are asymptomatic at the time of diagnosis.
Presentation of symptomatic CM1 differs with age. Children who are 2 years and younger most commonly present with oropharyngeal dysfunction (nearly 80%). Those between 3 and 5 years present with headache (57%) or scoliosis (38%). Children with holocord syringohydromyelia demonstrate altered pain, temperature, and vibratory sensation.
Uncommon presentations include hypersomnolence and sleep apnea. Valsalva-induced suboccipital headache (i.e., with coughing or sneezing), neck pain, and syncope are common in adults.
Natural History. The natural history of CM1 varies. Many patients remain asymptomatic. Some investigators believe that the degree of tonsillar ectopia in CM1 gradually increases with time and is associated with a greater likelihood of becoming symptomatic.
Outcomes of CM1-associated syringomyelia are likewise uncertain. Longitudinal studies show that a syrinx remains stable or decreases in size in nearly 90% of pediatric patients who have minimal neurologic symptoms. Others develop progressive scoliosis, spinal cord symptoms, or bulbar deficits. Such deficits are sometimes precipitated or exacerbated by relatively minor head or neck injury.
(36-18) 2D phase-contrast cine CSF flow study in Chiari I shows change in signal from dark to bright behind the cervical spinal cord, consistent with "pistoning" tonsillar pulsations.
Treatment Options. Like almost everything else about CM1, management is controversial. Asymptomatic tonsillar ectopia in the absence of an associated syrinx or scoliosis is usually not treated. Periodic surveillance of patients with documented hydrosyringomyelia is generally recommended, as 12% of syringes show increase in size and may require craniocervical decompression if symptoms worsen.
Treatment of symptomatic CM1 attempts to restore normal CSF fluid dynamics at the FM. Craniospinal CSF stroke volume, maximal cord displacement during the cardiac cycle, and evidence for collateralization of venous drainage around the FM correlate with favorable outcome following surgical decompression.
Imaging
General Features. The basion-opisthion line (BOL) is a line drawn from the tip of the clivus to the posterior rim of the FM (36-13). Measuring the distance from this line to the inferior margin of the cerebellar tonsils on sagittal MR defines tonsillar position.
Midline tonsillar descent 5 mm or more below the BOL—often considered diagnostic of Chiari 1—is by itself a poor criterion for definitive diagnosis. Tonsils 6 mm below the FM are common during the first decade of life. Almost 15% of normal patients have tonsils that lie 1-4 mm below the FM, and 0.5-1.0% have tonsils that project 5 mm into the upper cervical canal.
Great caution should be exercised in establishing a diagnosis of CM1, especially on the basis of borderline tonsillar ectopia alone. Unless (1) the tonsils appear compressed and pointed (peg-like) instead of gently rounded (36-15A), (2) the tonsillar

Congenital Malformations of the Skull and Brain
1178
folia are angled obliquely or inferiorly (instead of horizontally), and (3) the retrocerebellar CSF spaces at the FM/C1 level are effaced (36-15B), the diagnosis may not be warranted. Lowlying tonsils that retain their rounded shapes and are surrounded by normal-appearing CSF spaces are usually asymptomatic and of no diagnostic significance.
CT Findings. NECT scans may reveal a "crowded" FM and effaced retrotonsillar CSF space. Bone CT often demonstrates a combination of undersized, shallow posterior cranial fossa, short clivus, and CVJ assimilation anomalies. Look carefully for calvarial anomalies, as nearly 10% of patients with nonsyndromic single-suture craniosynostosis have CM1.
MR Findings. Sagittal T1 and T2 scans show "pointed" tonsils with more vertically oriented folia, obliterated retrocerebellar and premedullary subarachnoid spaces, and a "crowded" FM. The posterior fossa may appear normal or somewhat small with a short clivus and steeply angled straight sinus. In contrast to Chiari 2, the 4th ventricle usually displays a normal fastigium (dorsal point). In some cases, the inferior fourth ventricle is mildly elongated, and the nucleus gracilis—which demarcates the end of the obex and beginning of the central canal—can appear slightly low-lying (36-16).
The proximal cervical spinal cord should be carefully examined for the presence of hydrosyringomyelia. T2/FLAIR parenchymal hyperintensity without frank cyst formation may indicate a "pre-syrinx" state (36-15A).
The diameter of the central canal relative to the cord normally decreases significantly during the first few years of life. A CSFlike central cord cavity 3 mm or larger on axial scans is abnormal in older children and adults and should be considered a syrinx. Mean syrinx size in a large series of CM1 patients was nearly 8 mm in width and averaged nine vertebral levels in length.
Sagittal phase-contrast CSF flow studies show diminished or absent alternating bright (systolic) and dark (diastolic) signals behind the cervicomedullary junction. Any change in signal intensity of the cerebellar tonsils in cine mode suggests tonsillar pulsations (36-18).
Associated Abnormalities. Complete imaging evaluation in CM1 includes the brain, CVJ, and entire spine. Mild to moderate hydrocephalus is present in 10% of patients with CM1. Callosal dysgenesis is seen in 3.0% and absent septi pellucidi in 2.4% of cases. Other supratentorial anomalies are uncommon.
Hydrosyringomyelia is present in 10-20% of asymptomatic patients and 40-80% of symptomatic CM1 patients. Associated skeletal anomalies include a retroverted dens, clival-C2 body angle < 122°, hypoplastic C1 ring, Klippel-Feil anomaly, basilar invagination, platybasia, CVJ fusion anomalies, kyphosis, and/or scoliosis.
Differential Diagnosis
Congenital tonsillar descent (CM1) must be distinguished from normal variants (mild uncomplicated tonsillar ectopia). The most important pathologic differential diagnosis is acquired
tonsillar herniation caused by increased intracranial pressure or intracranial hypotension.
Increased intracranial pressure due to supratentorial mass effect with transmission of the pressure cone through the tentorial incisura can be easily distinguished from CM1. Signs of descending transtentorial herniation are present along with downward midbrain displacement. Tonsillar herniation in such cases is a secondary effect and should not be termed "acquired Chiari 1."
Intracranial hypotension shows a constellation of other findings besides inferiorly displaced tonsils. "Slumping" midbrain, enlarged pituitary gland, draping of the optic chiasm and hypothalamus over the dorsum sellae, subdural hematoma, engorged venous sinuses, and dura-arachnoid thickening and enhancement are typical abnormalities.
Mistaking intracranial hypotension for CM1 can have disastrous consequences, as surgical decompression may exacerbate brainstem "slumping."
Approximately 20% of patients with idiopathic intracranial hypertension exhibit cerebellar tonsillar ectopia ≥ 5 mm. Half have a peg-like tonsil configuration, and many have a low-lying obex. Looking for other signs of idiopathic intracranial hypertension (e.g., optic nerve head protrusion into the globe) is essential to avoid misdiagnosis as Chiari 1.
Other conditions that reduce posterior cranial fossa volume can also displace the tonsils below the foramen. Such causes of cranial constriction include craniosynostosis, achondroplasia, acromegaly, and Paget disease. Cranial "settling" from rheumatoid arthritis, osteogenesis imperfecta, hereditary connective tissue disorders, and occipitoatlantoaxial joint instability can also force the tonsils below the FM.
CHIARI 1 MALFORMATION
Clinical Issues
•Most common Chiari malformation
•Found in 3-4% of children on routine brain imaging
•Up to 50% asymptomatic
•Valsalva-induced suboccipital headache, neck pain
•Holocord hydrosyringomyelia
○Progressive scoliosis ± lower extremity pain
○Abnormalities of temperature, vibratory sensation
Imaging Findings
•General features
○Caudal tonsillar ectopia (≥ 5 mm below foramen magnum, FM)
○Pointed, peg-like tonsils with angled folia
○"Crowded" FM with effaced CSF spaces
○Diminished/absent CSF flow at posterior FM
○Syrinx in 10-20% asymptomatic, 40-80% symptomatic patients
•Differential diagnosis
○Normal "low-lying" tonsils (rounded, no disturbed CSF flow)
○Acquired herniation (elevated ICP, intracranial hypotension)
Posterior Fossa Malformations
1179
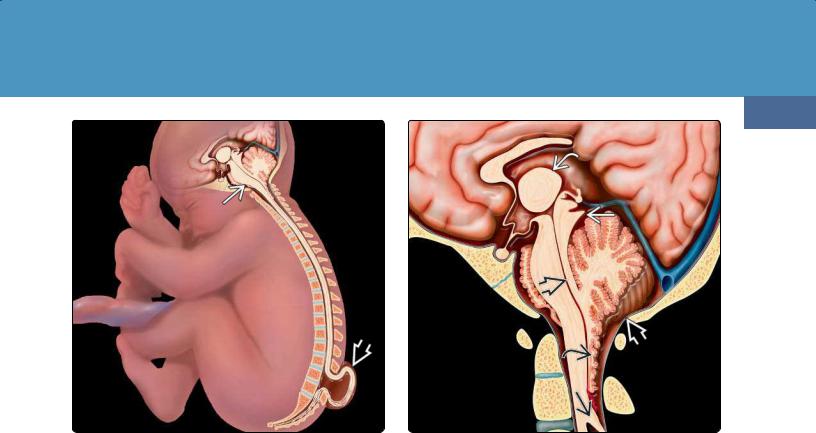
(36-19) Graphic depicts a fetus with Chiari 2 malformation with the spinal cord tethered into a myelomeningocele .
(36-20) Graphic shows CM2 with small posterior fossa , large massa intermedia , "beaked" tectum , callosal dysgenesis, elongated fourth ventricle with "cascade" of inferiorly displaced nodulus and choroid plexus, and medullary spur .
Chiari 2
Terminology and Definition
Chiari 2 malformation (CM2) is a complex hindbrain malformation that is almost always associated with myelodysplasia (myelomeningocele) (36-19).
Etiology
General Concepts. CM2 is a disorder of neural tube closure but also involves paraxial mesodermal abnormalities of the skull and spine. A number of steps are required for proper neural tube closure and formation of the focal expansions that subsequently form the cerebral vesicles and ventricles. Skeletal elements of both the skull and vertebral column become "modeled" around the neural tube.
Only if the posterior neuropore closes will the developing ventricles expand sufficiently for a normal-sized posterior fossa to form around the hindbrain. If this does not happen, the cerebellum develops in a small posterior fossa with abnormally low tentorial attachments. The growing cerebellum is squeezed cephalad through the tentorial incisura and stretched inferiorly through the foramen magnum (FM).
Genetics. Nearly half of all neural tube closure anomalies have mutations on the methylene-tetra-hydrofolate reductase gene (MTHFR). Maternal folate deficiency and teratogens such as anticonvulsants have been linked to increased risk of CM2.
Pathology
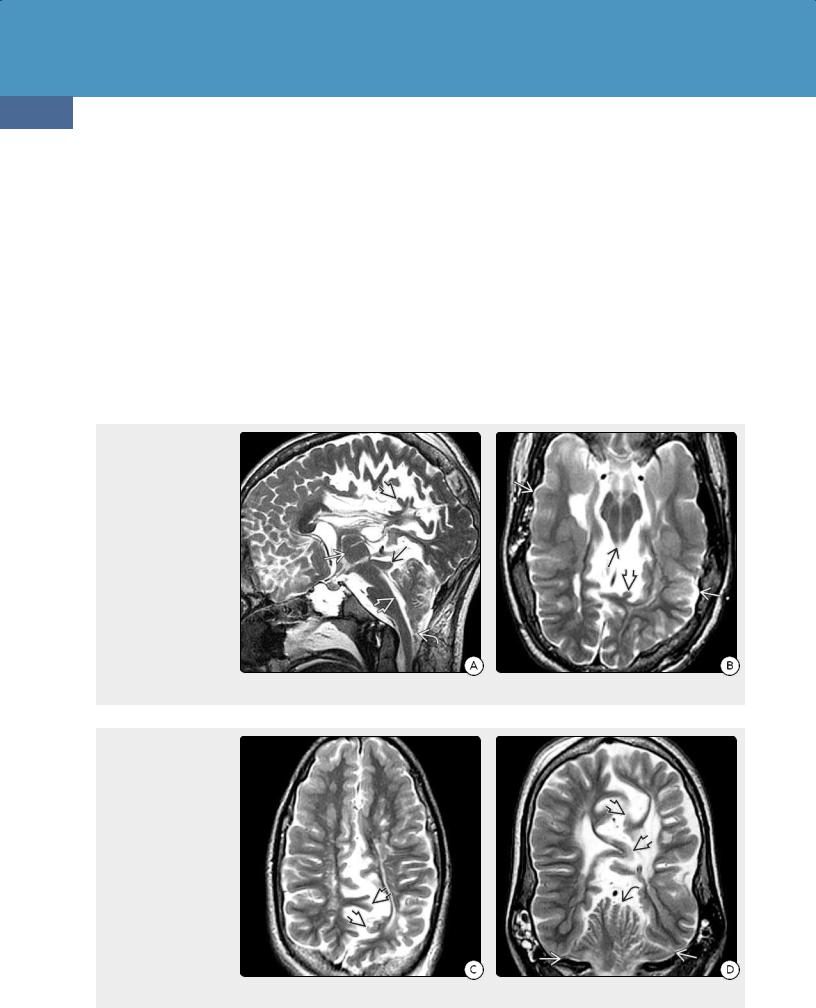
Grossly, a broad spectrum of findings can be present in CM2. Myelomeningocele and a small posterior fossa with concave clivus and petrous pyramids are virtually always present (3620). The cerebellar vermis (typically the nodulus) is displaced inferiorly along the dorsal aspect of the cervical spinal cord. The fourth ventricle, pons, and medulla are elongated and partially dislocated into the cervical spinal canal. The lower medulla may be kinked.
Unlike Chiari 1, supratentorial abnormalities are the rule in CM2, not the exception. Hydrocephalus is present in the majority of cases, and aqueductal stenosis is common. Corpus callosum dysgenesis and gray matter anomalies such as polymicrogyria and heterotopias are frequent (36-21) (36-22) (36-23) (36-24) (36-25) (36-26).
Clinical Issues
Epidemiology and Demographics. The overall prevalence of CM2 is 0.44 in 1,000 live births but has been decreasing with prophylactic maternal folate therapy. A dose of 4 mg per day reduces the risk of CM2 by at least 70%.
Presentation. CM2 is identified in utero with ultrasound or fetal screening for elevated α-fetoprotein. At birth, coexistent myelomeningocele and hydrocephalus are dominant clinical features in over 90% of cases. Lower cranial nerve deficits, apneic spells, and bulbar signs may be present. Lower extremity paralysis, sphincter dysfunction, and spasticity often develop later.
Treatment Options. Fetal repair of myelomeningocele is increasingly common and may reduce subsequent symptoms.
Congenital Malformations of the Skull and Brain
1180
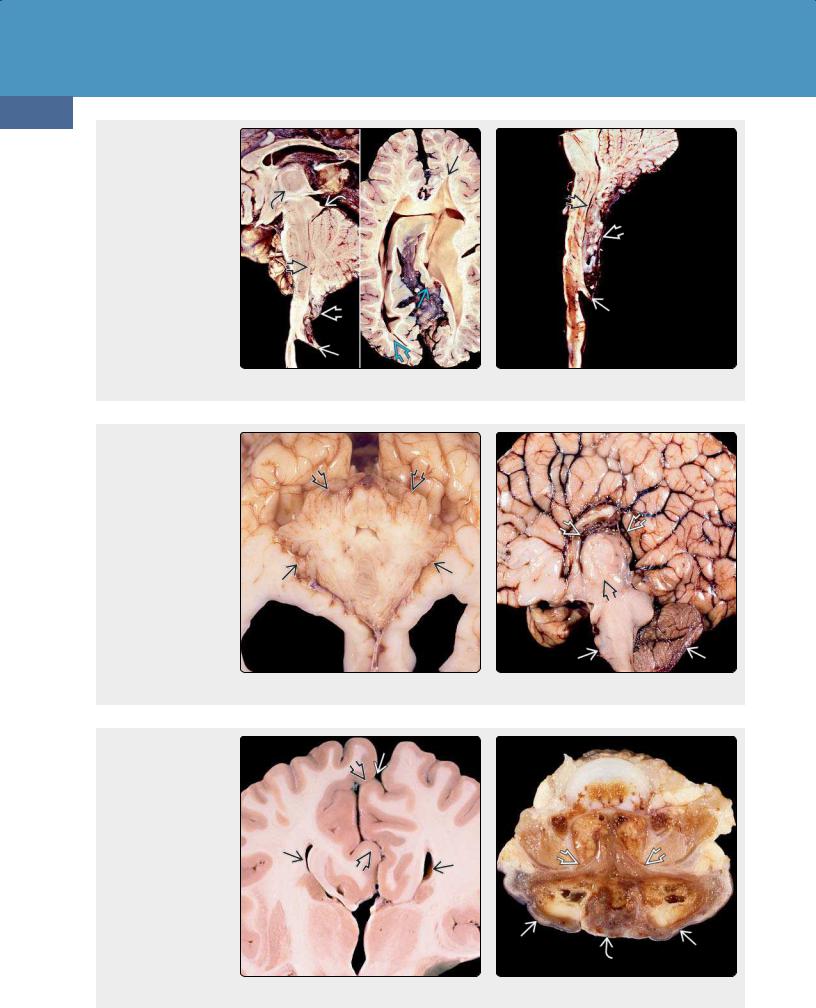
(36-21) (L) CM2 shows medullary spur , "cascade" of nodulus, CP behind medulla, elongated "soda straw" 4th ventricle , "beaked" tectum , large massa intermedia . (T. P. Naidich, MD.) (R) CM2 has stenogyria , heterotopic GM , "pointed" lateral ventricles . (E. T. Hedley-Whyte, MD.) (3622) CM2 shows medullary spur , "cascade" of nodulus, choroid plexus, elongated "strawlike" 4th ventricle . (R. Hewlett, MD.)
(36-23) Autopsy of CM2 shows a very small posterior fossa , cerebellar hemispheres "creeping" anteriorly around the medulla. (Courtesy R. Hewlett, MD.) (36-24) Autopsy of CM2 shows a very small posterior fossa , huge massa intermedia , corpus callosum dysgenesis with radiating gyri converging on a "high-riding" third ventricle that is open dorsally to the interhemispheric fissure. (Courtesy R. Hewlett, MD.)
(36-25) Coronal autopsy of CM2 shows corpus callosum agenesis with "Viking helmet" or "moose head" appearance to third, lateral ventricles. Note interdigitating gyri creating an irregular, "serrated" appearance to the interhemispheric fissure. (J. Townsend, MD.) (36-26) Axial view shows autopsied spine from CM2. Note wide dorsal dysraphism and myelomeningocele with exposed neural placode . (R. Hewlett, MD.)
Posterior Fossa Malformations
Surgical repair within 72 hours following delivery reduces mortality and morbidity from the open dysraphism.
Imaging
CM2 affects many regions of the skull, brain, and spine, so a variety of imaging abnormalities may be seen.
Skull and Dura. The calvarial vault forms from membranous bone. With failure of neural tube closure and absence of fetal brain distension, normal induction of the calvarial membranous plates does not occur. Disorganized collections of collagen fibers and deficient radial growth of the developing calvaria ensue, resulting in lacunar skull (i.e., Lückenschädel) (36-27). The skull defects ("craniofenestra") are caused by the mesenchymal abnormality and are not a consequence of increased intracranial pressure.
Focal calvarial thinning and a "scooped-out" appearance are typical imaging findings of lacunar skull. The calvaria appears
1181
thinned with numerous circular or oval lucent defects and shallow depressions. The craniofenestra diminish with age and typically resolve by 6 months, although some scalloping of the inner table often persists into adulthood.
A small, shallow, bony posterior fossa with low-lying transverse sinuses is almost always present in CM2. A large, "gaping" FM is common. Concave petrous temporal bones and a short concave clivus are often present (36-28).
Dural abnormalities are common. A widened, open, heartshaped tentorial incisura and thinned, hypoplastic, or fenestrated falx are frequent findings. The fenestrated falx allows gyri to cross the midline. Interdigitating gyri and the deficient falx result in the appearance of an irregular interhemispheric fissure on imaging studies (36-28C) (3629C).
Midbrain, Hindbrain, and Cerebellum. Hindbrain and cerebellum anomalies are a constant in CM2. The medulla and
(36-27) Autopsy case of lacunar (Lückenschädel) skull in CM2 shows multiple "scooped" out foci of thinned, almost translucent bone . (Courtesy R. Hewlett, MD.) (36-28A) NECT scan of a patient with CM2 shows a small posterior fossa with concave petrous ridges , scalloped inner table , no visible fourth ventricle, and "creeping" cerebellar hemispheres almost enveloping an elongated, inferiorly stretched medulla .
(36-28B) NECT scan in the same patient shows widely gaping, heartshaped incisura with "towering" cerebellum protruding superiorly and mild "beaking" of the tectum . (36-28C) More cephalad scan in the same patient shows the typical "serrated" appearance of the interhemispheric fissure due to the interdigitating gyri typically seen in CM2.
Congenital Malformations of the Skull and Brain
1182
cerebellar vermis (not the tonsils!) are displaced downward into the upper cervical canal for a variable distance. The inferiorly displaced cerebellar tissue is typically the nodulus, with variable contributions from the uvula and pyramid. A cervicomedullary "kink" with a "medullary spur" is common in the upper cervical canal but may lie as low as T1-4 in severe cases.
On sagittal T1 and T2 scans, the inferiorly displaced vermis, medulla, and choroid plexus form a "cascade" of tissue that protrudes downward through the gaping FM to lie behind the spinal cord. The superiorly herniated cerebellum may compress and deform the quadrigeminal plate, giving the appearance of a "beaked" tectum (36-29A) (36-29B).
In addition to the cephalocaudal displacement of posterior fossa contents, the cerebellar hemispheres often curve anteromedially around the brainstem. In severe cases, the pons and medulla appear nearly engulfed by the "creeping" cerebellum on axial imaging studies.
(36-29A) Sagittal image in a 13y patient demonstrates many features of Chiari 2, including small posterior fossa, elongated "soda straw" fourth ventricle, "cascade" of vermis/choroid plexus behind the medulla , "beaked" tectum , large massa intermedia , and multiple gyral malformations ("stenogyria") . (3629B) Axial T2WI shows "beaked" tectum , stenogyria , and scalloped calvaria .
(36-29C) Axial T2WI shows fenestrated falx with shortened interdigitating gyri , irregular "serrated" appearance to interhemispheric fissure. (36-29D) Coronal T2WI shows low-lying transverse sinuses with a very small posterior fossa, "towering" cerebellum that protrudes upward through the tentorial incisura, and interdigitating gyri , giving a "serrated" appearance to the interhemispheric fissure.
The cerebellar hemispheres and vermis are pushed upward through the incisura, giving the appearance of a "towering" cerebellum on coronal T1 and T2 scans (36-29D).
Ventricles. Abnormalities of the ventricles are present in over 90% of CM2 patients. The fourth ventricle is caudally displaced, typically lacks a fastigium (dorsal point), and appears thin and elongated ("soda straw" fourth ventricle). The third ventricle is often large and has a very prominent massa intermedia (36-29A).
The lateral ventricles vary in size and configuration. Hydrocephalus is almost always present at birth. The atria and occipital horns are often disproportionately enlarged ("colpocephaly"), suggesting the presence of callosal and forceps major dysgenesis.
Following shunting, the lateral ventricles frequently retain a serrated or scalloped appearance. A large CSF space between the occipital lobes often persists.

Posterior Fossa Malformations
Cerebral Hemispheres. Malformations of cortical development, such as polymicrogyria, contracted narrow gyri ("stenogyria") (36-29B), and heterotopic gray matter, are frequent associated findings.
Callosal dysgenesis is found in nearly two-thirds of all cases, and abnormalities of the fornices are also common.
Spine and Spinal Cord. Open spinal dysraphism with myelomeningocele is present in almost all cases of CM2.
Hydrosyringomyelia is seen in 50%.
Differential Diagnosis
The major differential diagnosis of CM2 is other Chiari malformations.
In Chiari 1, it is the tonsils (not the vermis) that are herniated inferiorly. Myelomeningocele is absent, and, other than being
1183
somewhat small, the posterior fossa and its contents appear relatively normal.
If findings of CM2 plus a low occipital or high cervical cephalocele are present, the diagnosis is Chiari 3.
A few cases of posteriorly angled odontoid, brainstem descent, and tonsillar ectopia without myelodysplasia have been described and are considered by some investigators as Chiari 1.5 (see below).
Severe chronic shunted congenital hydrocephalus may cause cerebellar herniation upward through the tentorial incisura, but brainstem descent and myelomeningocele are absent.
(36-30A) Sagittal T2WI in a 3d infant shows Chiari 3 with a cephalocele that contains herniated dysplastic brain and CSF in continuity with a lateral ventricle. (36-30B) Axial T1WI (L), T2WI (R) in the same patient show extension of the lateral ventricles into the cephalocele. (Courtesy G. Hedlund, DO.)
(36-31A) Chiari 3 is shown with extensive cranium bifidum extending from the occipital bone through the entire cervical spine . (36-31B)
Sagittal T1WI in the same patient with cranium bifidum and Chiari 3 shows an enormous meningocele sac with herniated, dysplasticappearing brain . The fourth ventricle is enlarged, elongated, and "tugged" toward the cephalocele. (Courtesy A. Illner, MD.)

Congenital Malformations of the Skull and Brain
1184
CHIARI 2 MALFORMATION
Pathoetiology
•Complex hindbrain malformation with myelomeningocele
○Posterior neuropore closure disorder
○Developing vesicles fail to expand
○Paraxial mesodermal abnormalities (skull, spine)
•Result = "too small" bony posterior fossa
Clinical Issues
•Prevalence reduced with maternal folate
•Myelomeningocele, hydrocephalus dominate clinical picture at birth
Imaging Findings
•Myelomeningocele (almost always)
•Lacunar skull
•Small posterior fossa
•Abnormal dura (gaping FM, heart-shaped incisura, fenestrated falx)
•Inferiorly displaced medulla, vermis lead to "cascade" of tissue
•Cervicomedullary "kink," medullary "spur"
•"Towering" and "creeping" cerebellum
•"Soda straw" fourth ventricle
•Prominent massa intermedia
•Hydrocephalus, shunted ventricles appear scalloped
•Callosal dysgenesis
•Stenogyria, gray matter heterotopias
Chiari 3
Terminology
Chiari 3 malformation (CM3) is the rarest of the Chiari malformations. CM3 consists of a small posterior fossa with a caudally displaced brainstem and variable herniation of meninges/posterior fossa contents through a low occipital or upper cervical bony defect.
Pathology
The cephalocele contains meninges together with variable amounts of brain tissue, vessels, and CSF spaces. The brain is often featureless, dysplastic-appearing, and disorganized with extensive gliosis and gray matter heterotopias.
Clinical Issues
The cephalocele in CM3 generally appears as a large skincovered, sac-like suboccipital mass protruding posteroinferiorly from the craniovertebral junction. Microcephaly is common, and, in extreme cases, the cephalocele exceeds the cranium in size (36-30).
Some cases are diagnosed with antenatal ultrasound. Other patients present at birth with bulbar and long tract signs, seizures, and developmental delay. Surgical mortality is high, and prognosis is generally poor because survivors usually have severe residual neurologic deficits.
Imaging
NECT scans show bony features similar to those seen in CM2, i.e., a small posterior cranial fossa, short scalloped clivus, lacunar skull, a defect in the ventral chondral portion of the supraoccipital bone, and low cranium bifidum that may extend inferiorly to involve much of the cervical spine (36-31).
MR best delineates sac contents, which often include dysplastic-appearing cerebellum and/or brainstem, as well as distorted CSF spaces and vessels. A deformed fourth and sometimes third ventricle can be partially found within the mass of herniated brain and meninges. Veins, dural sinuses, and even the basilar artery are sometimes "pulled" into the defect.
Differential Diagnosis
The differential diagnosis of CM3 includes isolated occipital cephalocele, iniencephaly, and syndromic occipital cephaloceles. Isolated occipital cephalocele lacks the typical intracranial features of CM2 and is not associated with cervical dysraphism.
Iniencephaly is an occipital cephalocele with extensive spinal dysraphism and fixed retroflexion of the neck ("stargazer" fetus). Syndromic occipital cephalocele occurs with other specific features (e.g., in Meckel-Gruber and Goldenhar-Gorlin syndromes).
CHIARI 3 MALFORMATION
Pathology
•Small posterior fossa
•Caudally displaced brainstem
•Low occipital or upper cervical bony defect
•Cephalocele with herniation of meninges, dysplastic brain, ventricles
Sac May Contain
•Meninges
•Dysplastic brain
•Deformed ventricle(s)
•Blood vessels (venous sinuses, arteries)
Chiari Variants
Some additions to the original Chiari classification have been proposed by neurosurgeons to account for hindbrain herniations that do not conform to the classic Chiari 1-3 definitions. Although these concepts are controversial and have not been universally adopted, radiologists should at least be familiar with them.
Chiari 0 Malformation
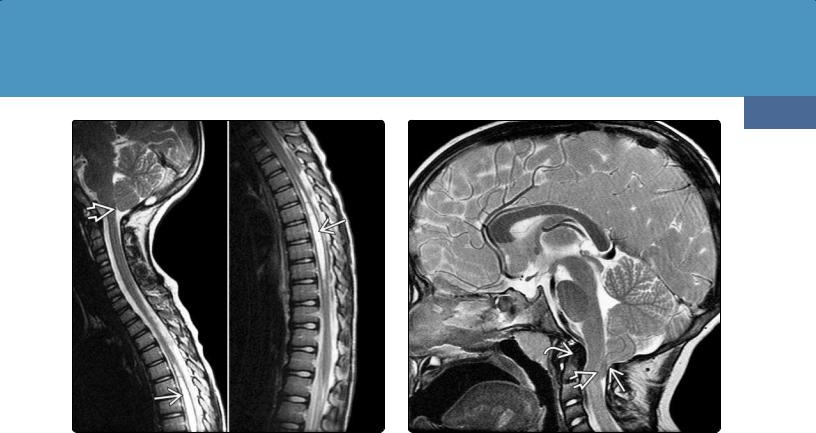
This variant consists of hydrosyringomyelia and foramen magnum (FM) "crowding." Chiari 0 differs from Chiari 1, as the cerebellar tonsils are normally positioned (i.e., either above or less than 3 mm below the FM) (36-32).
Chiari 0 patients are typically symptomatic (usually because of the syrinx). A smaller than normal posterior fossa (particularly
Posterior Fossa Malformations
1185
(36-32) Sagittal T2 scans show Chiari 0 malformation with thoracic syrinx . The cerebellar tonsil is rounded and in normal position, but the FM appears "crowded" posteriorly.
with a short clivus) and the presence of embryonic membranes at the foramen magnum may accentuate the craniovertebral junction obstruction.
Chiari 1.5 Malformation
Terminology. The term Chiari 1.5 malformation (CM1.5) has been coined by neurosurgeons to designate a "complex Chiari" malformation in which cerebellar tonsillar herniation is complicated by other abnormalities (e.g., caudally displaced brainstem and fourth ventricle and/or cervicomedullary "kink"). CM1.5 differs from classic CM1 in that caudal descent of the brainstem is present, and tonsillar herniation is typically more severe. CM1.5 differs from CM2, as myelomeningocele is absent.
Clinical Issues. Recent large series show that cases fulfilling the imaging criteria for CM1.5 represent approximately 22% of all patients 16 years or younger referred for surgical management of Chiari-related malformations.
Age at diagnosis is usually younger compared with CM1 although no single sign or symptom is peculiar to CM1.5. The most frequent symptom is headache (often Valsalva induced). Progressive scoliosis and syrinx-related symptoms such as extremity paresthesias are common.
Symptomatic patients with "classic" CM1 often require only suboccipital decompression (with or without duraplasty), whereas patients with "complex" CM1.5 abnormalities may require other additional interventions, such as transoral odontoid resection and occipitocervical fusion. Outcomes are similar, suggesting that CM1.5 is just a more severe subtype of CM1 rather than a unique type of Chiari malformation.
(36-33) Sagittal T2WI in a 6y girl with Chiari 1.5 malformation shows retroflexed odontoid , tonsillar herniation , crowded FM, and low-lying nucleus gracilis . The clival-C2 body angle is normal (i.e., > 122°).
Imaging. In addition to tonsillar ectopia (see above), patients with CM1.5 demonstrate several other significant imaging abnormalities. The major finding that differentiates CM1.5 from CM1 is the presence of brainstem herniation through the FM (hence the term "Chiari 1.5 malformation"). Elongation/caudal displacement of the brainstem and fourth ventricle and displacement of the obex below the FM are common (36-33). FM "crowding" with a medullary "kink" or "bulb" is often present in addition to the inferiorly displaced tonsils.
Bony abnormalities are common in CM1.5. These include a "retroflexed" odontoid, abnormal clival-cervical angle, occipitalization of the atlas, basilar invagination with odontoid compression of the brainstem, and scoliosis. Syringomyelia is present in 50% of cases, but spina bifida and myelodysplasia are absent.
Differential Diagnosis. The major differential diagnosis of CM1.5 is CM1. As management strategies differ, the distinction is important. Although both CM1.5 and CM1 share common features, such as tonsillar descent and bony anomalies, caudal brainstem descent distinguishes the two malformations.
Chiari 4 Malformation
The terms "primary cerebellar agenesis" or "severe cerebellar hypoplasia" should be used instead of "Chiari 4 malformation." The posterior fossa is normal in size and mostly filled with CSF. The pons is small and appears flat. There is no myelomeningocele, and the intracranial features of Chiari 2 are absent.