

Соли d-элементов
Фосфаты, карбонаты, силикаты, сульфиды и фториды d-металлов малорастворимы, в отличие от их сульфатов, нитратов, перхлоратов и галидов. Исключение составляют серебро и медь в ст.ок. (+1),галидыкоторых (кроме как раз фторидов) малорастворимы (?).
Также малорастворимы хлориды платины и аналоговванадия, хрома и марганца в ст.ок. (+2), которые являютсякластерами(?). Указанные элементы склонны к сохранению связи M− M в соединениях снизкойстепенью окисления, т.к. имеют большое число неспаренных валентных электронов и, как следствие, образуют прочную межатомную связь в металлической решетке. Например, энергия атомизации платины 556 кДж/моль, а у палладия, не образующего кластеры, всего 381 кДж/моль.
Комплексные соединения
Мы присутствуем при процессе диффузии представлений химии комплексных соединений во все решительно области чистой и прикладной химии, а также биохимии. А.А. Гринберг
Конфигурация комплексов.Для большинства d-элементов (от начала декад до подгруппы кобальта) характерно к.ч.=6 (октаэдр) и выше (до 9 у La). Однако по мере увеличения числа валентных электронов и уменьшения количества свободных орбиталей к концу декад к.ч. снижается до 4 и даже до 2 (при ст.ок. (+1) у ц.а.).
Отметим, что если электронная конфигурация иона – d8(Pd2+ , Pt2+ , Au3+ ), то при к.ч.=4 и достаточносильномполе лигандов (L)энергетическивыгоднее не тетраэдрическое, аквадратноеокружение (рис. 4).
Катионные комплексы.Аквакомплексыобразуются при растворении в воде солей, в которых d-элемент является катионом. Их состав - Э(H2O)n6+ . (Лишь у Pd, Cu и Zn к.ч.=4). Молекулы воды, являясь лигандамисреднегополя, образуют, как правило,
|
высокоспиновыекомплексы [3]. |
|
|
|
|
|
dx2-y2 |
Поэтому |
прочность, в |
а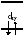
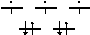
 )
б)частности,октаэдрическихаквакомплексовувеличивается
)
б)частности,октаэдрическихаквакомплексовувеличивается
dxy dxz dyz при заполнении электронамимежосевыхорбиталей (d ),ε поскольку ониболеенизкипо
dz2 энергии (рис. 5). При этом ростет
dx2-y2 dz2 энергиястабилизациикристаллическимполемлигандов (ЭСКП). Заселение dε по
|
dxz dyz Рис.4. Энергетическая диаграмма d-орбиталей при конфигурации d8 в тетраэдрическом (а) и в квадратном (б) окружении лигандов |
одномуе, происходит при переходе от Sc к V (рис. 5а); а повторому- от Mn к Ni. При этих переходах и растет устойчивость аквакомплексов. Напротив, их |
прочность снижаетсяпри заполненииосевыхорбиталей (dγ ), обладающих более высокой энергией, т.е. при переходе от V к Mn (рис. 5б) и от Ni к Zn.
Аммиачныекомплексыобычно получают действием аммиака на водные растворы солей, иногда в присутствии NH4Cl (зачем?). Их устойчивость в декаде растет до Cu2+ в соответствии с изменением кислотности по Льюису (лишь к Zn2+ снижается, поскольку при конфигурации d10 не происходит упрочнения комплексов за счет ЭСКП).
Так, для IIIБ – VБ групп аммиачные комплексы не получены; для подгрупп Cr и Mn синтезированы лишь в неводнойсреде, а водой разрушаются. Комплексы Co (2+ и
а) б)
d γ
(dx2
– y2,
dz2)
dε
(dxy,
dxz,
dyz)
γ
(dx2
– y2,
dz2)
dε
(dxy,
dxz,
dyz)
Рис.5. Энергетическая диаграмма d-орбиталей в октаэдрическом окружении лигандов – молекул воды а) для V(II); б) для Mn(II).
гораздо менее прочные в случае Fe2+ ) существуют в водных растворах, но только при большомизбыткеаммиака.
Аммиачные комплексы Fe3+еще менее устойчивы, вследствие достаточнонизкойрастворимости Fe(OH)3(ПР= 4⋅10−38 ), а [Co(NH3)6 ]3+(конфигурация d6εd0γ ), напротив,гораздопрочнее, чем [Co(NH3)6 ]2+(конфигурация d3εd2γ ): не разрушается даже приподкислении.
В подгруппах устойчивость аммиачных комплексов изменяется неоднозначно: от Ni к Pt – растет, в IБ группе – снижается, а в подгруппе Zn наименее стоек комплекс кадмия (что соответствует наименьшей сумме: I1 + I2 ).
Анионные комплексы.Анионные комплексы d-элементы образуют почти со всеми лигандами. Известны оксо-, гидроксо-, циано-, роданокомплексы [8], сульфидные (K2WS4 ), тиосульфатные (Na3[Ag(S2O3) ]2 ), карбонильные, галидные и пероксокомплексы. Последние можно синтезировать как в кислой среде:
Cr2O27− + H2O2 + H+ → H2CrO6(точнее CrO(O2 )2 ⋅ H2O – синего цвета),
так и в щелочной:
VO34− + H2O2 → H2O+ [VO2 (O2 )2 ]3−(желтого цвета).
Кроме того, d-элементы могут давать двойные соли:
M![]() H2O
(где ЭII– Zn ,2+
Mn ,2+
Ni ,2+ Cu
,2+
Fe2+
),
H2O
(где ЭII– Zn ,2+
Mn ,2+
Ni ,2+ Cu
,2+
Fe2+
),
M130ЭIII (SO4 )2 ⋅12H2O (где ЭIII– Sc3+и его аналоги, Cr ,3+ Fe3+ , Co ) –3+
такого рода двойные соли называют квасцами [8].
Более точная формула, например, калиевых квасцов (как показал рентгеноструктурный анализ): K(H2O)6 Э(H2O)6 (SO4 )2 ; т.е. они, как и обычные кристаллогидраты, например, CrCl3 ⋅ 6H O2 , вследствие координационной насыщенности ц.а. имеютионнуюрешетку и потому быстро растворяются в воде. (А безводные соли d-металлов часто представляют собойполимерыс ковалентными связями (например, CrCl3 имеет слоистую решетку) и, как следствие, растворяются очень медленно.)
Из галидныхкомплексов для элементов начала декад устойчивы лишь фторидные, причем в подгруппах значение к.ч. закономерно растет за счет увеличения r и валентности Э, а также вклада d- и f-сжатий в повышение прочности связи Э− L :
[TiF6 ]2− , но [ZrF8 ]4−и [HfF8 ]4−; [CrF6 ]0 , но [WF8 ]2−и [MoF8 ]2− .
Для скандия и его аналогов (вследствие их высокой металличности) устойчивость даже фторидных комплексов невелика1. Причем, если [ScF4 ]−можно получить в водном растворе, действуя на ScF3 любымщелочным фторидом, то для болееметалличноголантана комплекс [LaF6 ]3−образуется лишь при сплавлении LaF3 с CsF.
Начиная с ниобия и тантала, а в первой декаде – с Mn , кроме фторидных синтезированы и хлоридные комплексы состава: [ЭCl6 ]n−. Однако, например, для марганца фторидные комплексы более прочны – получены даже для Mn(II): [MnF4 ]2−, а хлоридные – лишь для Mn(IV)131.
Устойчивее фторидные комплексы и для Fe(III). Так, значение pKнест.[FeF6 ]3−равно
16,1, а для [FeCl4 ]−всего – 0,85. Для аналогов железа фторидные комплексы настолько прочны, что образуются при гидролизе (как и в случае фторидов бора и кремния):
ЭF4 + H2O→ ЭO2 + H2[ЭF6 ].
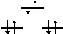
Напротив, для аналогов Co (несмотря на бóльшую величину расщепления d-орбиталей в поле фторид-ионов (рис. 6)) более характерны хлоридныекомплексы иззаусиленияπ-дативных132свойств элементов в подгруппах. (В результате возрастания подвижности d-электронных пар вследствие все большего d-экранирования ядра). Причем, в хлоридном КС за счет d6ε -конфигурации стабилизируется ст.ок. (+3) даже у иридия:
Na2[IrCl6 ]+ NaI→ Na3[IrCl6 ]+ I2 .
Начиная с подгруппы никеля, образуются бромидные и иодидные КС, а фторидные становятся неустойчивыми (?), особенно для Pt(IV).
Поскольку с увеличением номера периода растет вклад π-дативного взаимодействия, то комплексы [PtCl6 ]2− , [AuCl4 ]−и [HgI4 ]2−очень прочны – не разрушаются даже при действии нитрата серебра, а образуют осадки, например, Ag2[HgI4 ]. (Это вещество желтого цвета при нагревании краснеет, поэтому его можно использовать в качестве термокраски.)
а) б)


Рис.6. Энергетическая диаграммы [IrГ6]3- а) Г- - фторид-ионы б) Г- - хлоридионы.
Хлоридный комплекс платины(II) менее устойчив, чем платины(IV) (?), и уже при 200С дисмутирует (при этом красный раствор желтеет):
[PtCl4 ]2− → Pt+ [PtCl6 ]2− .
Карбонильные и цианидные КС.Частицы СО и CN−изоэлектронныи обе являются лигандами сильного поляσ-донорного-π-акцепторного типа. Как следствие, наиболее прочные комплексы они образуют с d-металлами, имеющими НЭП на валентном уровне.
Однако СО, в отличие от отрицательнозаряженного CN ,− какσ-донор слабее, а какπ-акцепторсильнее, поэтому более прочные комплексы, чем CN−, образует с элементами внулевойст.ок. (в которой М имеет максимальное число НЭП):
Ni+ CO⎯⎯→t0 Ni (CO)4 .
80 C
Эффективный заряд (δ) на атоме металла в таких КСположителен, в частности, в комплексе [Cr(CO) ]6δ(Cr)= +0,4 , но это меньше, чем, например, в CrCl (3 δ(Cr)= +1,2).
Получены также карбонилы, в которых ц.а. имеет отрицательнуюст.ок.: Na[Mn(CO) ]5 .
Значения к.ч. в карбонильных КС определяются числом свободных валентных орбиталей (включая и р-подуровень) при условии максимальногоспаривания электронов133, поэтому каждой электронной конфигурации ц.а. отвечает свойсоставкомплекса, например: Cr(CO)6 , Fe(CO)5 , Ni(CO)4 .
Если число электронов на валентном уровне ц.а. нечетное, то за счет электрона, оставшегося неспаренным(в поле действия лигандов), образуется двуядерное соединениекластерноготипа, т.е., напоминаем, имеющее связь M− M:
V2 (CO)12 , Mn2 (CO)10 , Co2 (CO)8 .
Таким образом, все указанные карбонильные комплексы обладают диамагнитными свойствами.
Двуядерные карбонилы получены и для соединений с четнымчислом валентных электронов, например, Fe2 (CO)9 :

CO
Синтезированы также три-, тетра- и даже гексаядерные соединения, например: Rh6 (CO)16 .
Карбонильные комплексы токсичны, растворимы в органических жидкостях, но не в воде (поэтому легко могут быть отделены от растворимых в ней примесей). Кроме того, моноядерные соединения довольно легко отделить от двуядерных, т.к. первые при об.у. - жидкости (т.пл. ≈ −200 C ), а вторые – твердые, хотя и легкоплавкие вещества (т.пл.≈ 50−1500 C).
При термическом разложении карбонилов (т.разл.(Ni (CO)4 )= 1800 C) образуются металлы (в порошкообразном состоянии) и оксид углерода(II). Так что реакции образования карбонилов можно использовать для получения особо чистых М, причем с регенерацией исходного реагента (СО).
Кроме того, карбонилы применяют для нанесения металлических покрытий на поверхность изделий сложной формы, а также в качестве катализаторов, т.к. они могут вступать в реакции замещения:
Cr(CO)6 + 3PF3 = Cr(PF3)3(CO)3 + 3CO,
а также участвовать в окислительно-восстановительных процессах:
Fe(CO)5 + Br2 → [Fe(CO)4 ]Br+ CO,
Fe(CO)5 + Na⎯⎯(NH⎯3)⎯ж → CO+ Na2[Fe(CO)4 ].
(При действии воды на последнюю соль образуется кислота H2Fe(CO)4 .)
Некоторые карбонилы, например, Fe(CO)5и Ni(CO)4 , настолько химически активны, что дают взрывчатые смеси с воздухом.
Для ряда d-элементов получены и цианидные КС в нулевойст.ок.: K4[Э(CN)4 ], где
Э = Co, Ni, Pd (восстановлением комплекса K2[Э(CN)4 ] с помощью калия в жидком аммиаке). Но они являются сильными восстановителями:
K 4[Э(CN)4 ]+ H2O→ K2[Э(CN)4 ]+ H2 + KOH.
Устойчивые же цианидные КС образуются с элементами в положительнойст.ок.
Как результаттого, что CN -− лигандсильногополя, а значит, вызывает значительное расщепление d-орбиталей по энергии, является следующее:
а). Образуется тетраэдрический комплекс [Mn(CN)4 ]2−– единственный
низкоспиновыйдля Mn(II);
б). [Ni(CN)4 ]2−– единственный для Ni комплексквадратнойформы;
в). Ru и Os стабилизируются в нехарактернойдля них ст.ок. (+2) за счет
устойчивой конфигурации d6εd0γв октаэдрическом поле лигандов CN .− Однако Fe(II)легчепереходит в Fe(III) при замене аквалигандов цианид-ионами (значение E0 при этом уменьшается с 0,77 до 0,36 В), что объясняется особой прочностью [Fe(CN)6 ]3− .
г). Резко повышаются восстановительныесвойства Co(II), как результат формированиянизкоспиновогокомплекса [Co(CN)6 ]4−с электронной конфигурацией d6εd1γ . В отсутствие окислителя данный комплексдимеризуется(с отщеплением KCN),
давая кластер: K6[Co2 (CN) ]10 , чтопонижаетэнергию системы.
Кроме того, являясь лигандом σ-донорного-π-акцепторного типа, CN−образует комплексы, которые на несколько порядков прочнее аммиачных или фторидных (для элементовсерединыиконцадекад). Это используется, например, в гидрометаллургии серебра и золота (значения Kd(Э(CN)2− ) равны соответственно 8⋅10−22и 5⋅10−39 ):
Э + KCN+ O2 + H2O→ K[Э(CN)2 ]+ KOH .
Из полученных растворов благородные металлы затем восстанавливают, используя, например, цинковую пыль.