

Патанатомия (Пособие для резидентуры)
.pdfБОЛЕЗНИ СИСТЕМЫ КРОВИ |
9 |
|
|
тов Ig. Другой пример – лимфома Ходжкина, которая часто ассоциируется с лихорадкой, обусловленной высвобождением воспалительных цитокинов.
Классификация лимфоидных неоплазий достаточно противоречива, однако успехи в разработке молекулярных методов диагностики позволили существенно улучшить эту ситуацию.
Согласно классификации Всемирной организации здравоохранения, основанной на морфологических, иммунофенотипических, генотипических и клинических признаках, лимфоидные неоплазии разделены на 5 категорий соответственно их клеточному происхождению (табл. 9.1):
пре-В-клеточные неоплазии (без созревания В-клеток);
периферические В-клеточные неоплазии (с дифференцировкой клеток);
пре-Т-клеточные неоплазии (без созревания Т-клеток);
периферические Т-клеточные и NK-клеточные неоплазии (с дифференцировкой клеток);
лимфома Ходжкина (с клетками Рид-Штернберга и их вариантами).
Таблица 9.1. Классификация лимфоидных неоплазий ВОЗ /7/.
Пре-В-клеточные неоплазии |
В-клеточная острая лимфобластная лейкемия/лимфома |
|||
|
|
|
||
|
|
Хроническая лимфоцитарная лейкемия/мелкоклеточная лим- |
||
|
|
фоцитарная лимфома |
||
|
|
В-клеточная пролимфоцитарная лейкемия |
||
|
|
Лимфоплазмоцитарная лимфома |
||
|
|
Лимфома из клеток маргинальной зоны (лимфома из клеток |
||
Периферические В-клеточные |
маргинальной зоны селезенки и лимфоузлов, экстранодальная |
|||
лимфома из клеток маргинальной зоны) |
||||
неоплазии |
|
|||
|
Лимфома из клеток мантийной зоны |
|||
|
|
|||
|
|
Фолликулярная лимфома |
||
|
|
Волосатоклеточный лейкоз |
||
|
|
Множественная миелома/солитарная плазмоцитома |
||
|
|
Диффузная В-крупноклеточнаяЦлимфома |
||
|
|
Лимфома Беркитта |
||
|
|
|||
Пре-Т-клеточные неоплазии |
Т-клеточная острая лимфобластная лейкемия/лимфома |
|||
|
|
|
||
|
|
Т-клеточная пролимфоцитарная лейкемия |
||
|
|
Крупноклеточная гранулярная лимфоцитарная лейкемия |
||
|
|
Грибовидный микоз/синдром Сезари |
||
|
|
Периферическая Т-клеточная лимфома неуточненная |
||
|
|
Анапластическая крупноклеточная лимфома |
||
Периферические Т-клеточные |
Ангиоиммунобластная Т-клеточная лимфома |
|||
и NK-клеточные неоплазии |
Т-клеточная лимфома, ассоциированная с энтеропатией |
|||
|
|
Э |
||
|
|
Панникулитоподобная Т-клеточная лимфома |
||
|
|
Печеночно-селезеночная γδ-Т-клеточная лимфома |
||
|
|
Т-клеточная лейкемия/лимфома взрослых |
||
|
|
Экстранодальная T/NK-клеточная лимфома |
||
|
|
NK-клеточная лейкемия |
||
|
|
|
||
ГЛимфома Ходжкина |
|
Нодулярный вариант с лимфоидном преобладанием |
||
|
Классические типы: |
|||
|
|
Нодулярный склероз; |
||
|
|
Смешанно-клеточный вариант; |
||
|
|
|
лимфоидное преобладание (преобладание лимфоидной |
|
|
|
|
ткани); |
|
|
|
лимфоидное истощение (истощение лимфоидной ткани). |
||
163

ОБЩАЯ ПАТОЛОГИЧЕСКАЯ АНАТОМИЯ
Прежде чем описывать представленные в классификации ВОЗ неоплазии, перечислим некоторые важные положения общего характера, касающиеся лимфоидных неоплазий:
лимфоидную неоплазию можно заподозрить на основании клинических признаков, однако для постановки диагноза необходимо гистологическое исследование лимфоузлов или других пораженных тканей;
в большинстве лимфоидных неоплазий перестройка генов антигенных рецепторов предшествует трансформации, поэтому все дочерние клетки, образовавшиеся из злокачественной клеткипредшественника, имеют те же конфигурацию и последовательностьЦгенов антигенных рецепторов и синтезируют идентичные рецепторные белки (Ig или Т-клеточные рецепторы). В отличие от этого при нормальном иммунном ответе образуются поликлональные популяции лимфоцитов, экспрессирующие различные антигенные рецепторы. Анализ генов антигенных рецепторов и их белковых продуктов позволяет отличить реактивную (поликлональную) лимфоидную пролиферацию от злокачественной (моноклональной). Кроме того, в результате перестройки генов антигенных рецепторов образуется уникальная последовательность ДНК – высокоспецифичный клональный маркер, который можно использовать для определения небольшого количества оставшихся после терапии злокачественных лимфоидных клеток;
лимфоидные неоплазии в основном имеют В-клеточное происхождение (85–90%). Оставшиеся неоплазии, за исключением редко встречающихся NK-клеточных неоплазий, относят к Т- клеточным. В большинствеЭслучаев опухолевые клетки лимфоидных неоплазий напоминают В- или Т-клетки различных стадий дифференцировки. Это используют для классификации лимфоидных неоплазий;
лимфоидные неоплазии часто ассоциируются с иммунными нарушениями: утратой иммунитета (восприимчивостью к инфекциям) или срывом толерантности (аутоиммунитетом). Иногда у одного и того же пациента присутствуют оба нарушения. С другой стороны, индивиды с врожденным или приобретенным иммунодефицитом уже имеют повышенный риск развития некоторых лимфоидных неоплазий, особенно вызываемых онкогенными вирусами;
функции неопластических В- и Т-клеток похожи на функции их нормальных аналогов. Как и нормальныеГлимфоциты, неопластические В- и Т-клетки мигрируют в определенные участки тканей, обусловливая характерную локализацию злокачественного процесса. Например, клетки фолликулярных лимфом мигрируют в герминативные центры лимфоузлов, тогда как клетки Т- клеточных лимфом локализуются в коже. Миграцию неопластических лимфоидных клеток, как и их нормальных аналогов, регулируют определенные молекулы адгезии и рецепторы хемокинов. Неопластические лимфоидные В- и Т-клетки рециркулируют, распространяясь по лимфатическим и кровеносным сосудам по всему организму, поэтому многие лимфоидные опухоли ко времени постановки диагноза уже диссеминированы. Исключением являются лимфомы из клеток маргинальной зоны, локализация которых часто ограничена очагом хронического воспаления, и лимфома Ходжкина, распространение которой иногда ограничено одной группой лимфоузлов;
распространение лимфомы Ходжкина происходит более предсказуемым образом, чем ранняя диссеминация большинства форм неходжкинских лимфом. Определение стадии развития лимфомы имеет прогностическое значение, особенно в случае лимфомы Ходжкина /7/.
Пре-В- и пре-Т-клеточные неоплазии
Острая лимфобластная лейкемия/лимфома. Опухолевые клетки при острой лимфо-бластной лейкемии/лимфоме (ОЛЛ) представляют собой незрелые В-клетки (пре-В-клетки) или Т-клетки (пре- Т-клетки), называемые лимфобластами. Около 85% всех ОЛЛ – это В-кле-точная ОЛЛ (В-ОЛЛ), в ти-
164
БОЛЕЗНИ СИСТЕМЫ КРОВИ |
9 |
|
|
пичных случаях проявляющаяся как детская острая лейкемия. Реже встречающаяся Т-клеточная ОЛЛ (Т-ОЛЛ) наблюдается преимущественно у подростков в виде тимусной лимфомы. Клиническая картина В-ОЛЛ и Т-ОЛЛ близка: например, В-ОЛЛ может принимать необычную форму опухолевой массы в коже или костях, а Т-ОЛЛ во многих случаях изначально или со временем дает клиническую картину лейкемии. Из-за морфологического и клинического сходства различные формы ОЛЛ рассмотрены вместе /6/.
ОЛЛ – наиболее частая злокачественная опухоль у детей. Заболевание чаще встречается у мальчи-
ОЛЛ наблюдается в подростковом возрасте, когда тимус достигает максимального размера. В-ОЛЛ и Т-ОЛЛ у взрослых регистрируют реже.
ков. Пик заболеваемости приходится на 3-летний возраст, возможно потому, что количество пре-В- клеток в нормальном костном мозге больше всего именноЦв этом возрасте. Пик заболеваемости Т-
При лейкемии костный мозг отличается гиперклеточностью и обилием лимфобластов, плотные скопления которых замещают нормальные элементы костного мозга. Медиастинальные опухолевые массы в тимусе встречаются в 50–70% случаев Т-ОЛЛ. В этой ситуации более вероятна связь с лимфаденопатией и спленомегалией. И при В-ОЛЛ, и при Т-ОЛЛ опухолевые клетки имеют скудную агранулярную базофильную цитоплазму и более крупные ядра, чем у малых лимфоцитов. Ядерный хроматин имеет тонкую точечную структуру, ядрышки либо отсутствуют, либо незаметны. Во многих случаях ядерная мембрана глубоко разделена, что придает ей инвагинированный вид. В соответствии
с агрессивным клиническим течениемЭв клетках высока частота митоза. Как и в случае других быстро-
растущих опухолей, рассеянные в опухоли макрофаги, поглотившие апоптозные опухолевые клетки, могут создавать картину «звездного неба». Лейкемическая инфильтрация выражена наиболее резко в костном мозге, селезенке, лимфатических узлах, лимфатическом аппарате ЖКТ, почках и вилочковой железы. Костный мозг трубчатых и губчатых костей малиново-красный, сочный. Селезенка резко увеличивается, становится сочной и красной, рисунок ее стерт. Значительно увеличиваются лимфатические узлы средостения и брыжейки, на разрезе их ткань бело-розовая, сочная. Такой же вид имеет
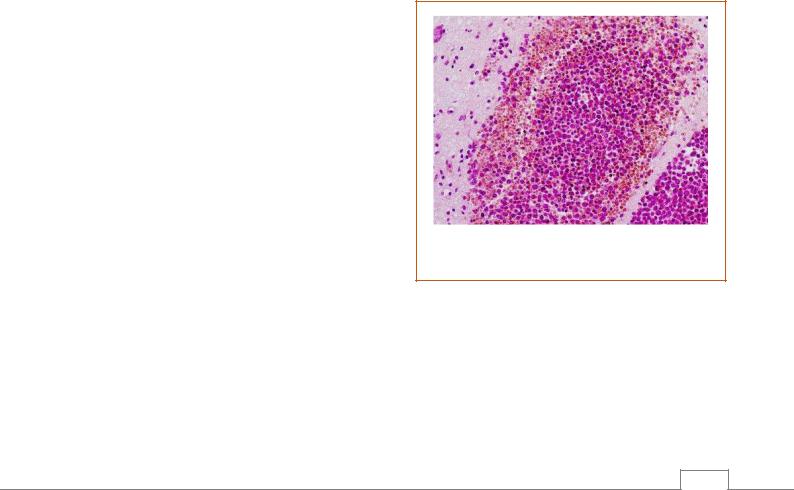
вилочковая железа, которая достигает иногда гигантских размеров. Лейкозные инфильтраты состоят из лимфобластов относящихся к Т-системе лимфопоэзаГ, для которых характерно наличие вокруг ядра гликогена. Выражением прогрессии лейкоза являются метастатические лимфобластные инфильтраты, которые особенно часто встречаются в оболочках и веществе головного и спинного мозга, что называют нейролейкемией (рис. 9.2).
Вследствие неодинаковой реакции опухолей на химиотерапию ОЛЛ необходимо отличать от острой миелоидной лейкемии (ОМЛ) – неоплазии, состоящей из незрелых миелоидных клеток, но которая может иметь идентичные признаки и симптомы.
По сравнению с миелобластами лимфобласты имеют более конденсированный ядерный хроматин, менее заметные ядрышки и меньшее количество цитоплазмы, в которой обычно отсутствуют гранулы. Однако эти морфологические различия неабсолютны и окончательный диагноз базируется на окрашивании с помощью антител, специфичных для антигенов В- и Т-клеток. Гистохимический метод позволяет установить, что в отличие от миелобластов лимфобласты дают отрицательную реакцию на миелопероксидазу и часто содержат ШИК-поло-жительный цитоплазматический материал.
В отличие, от всех других лейкозов, т.е. нелимфобластных, ОЛЛ характеризуется по нижеприведенным дифференциально-диагностическим особенностям:
165

ОБЩАЯ ПАТОЛОГИЧЕСКАЯ АНАТОМИЯ
эти лейкозы берут свое начало от лимфобластов;
около 80% случаев встречается, в детском возрасте, в основном до 5 лет;
при этом в лейкозных клетках выявляется терминальная дезокситрансфераза (ядерный фермент), который при острых нелимфобластных лейкозах не встречается;
в лейкозных клетках могут наблюдаться, хромосомные аномалии различного характера;
увеличение всех лимфатических узлов приводит к генерализации лимфоденоме-галии. При
остром нелимфобластном лейкозе увеличение лимфатических узлов обычно не наблюдается /7/.
сходны. В обоих случаях накопление неопластических бластов в костном мозге подавляет нормальный гемопоэз в результате физического сдавления, конкуренции за факторы роста и других малоизученных механизмов. Общие проявления и признаки ОЛЛ и ОМЛ:
Хотя ОЛЛ и ОМЛ генетически и иммунофенотипическиЦразличаются, клинически они очень
резкое, бурное начало с появлением первых симптомов в течение нескольких дней или недель;
симптомы, связанные с угнетением функции костного мозга, включая утомляемость, обусловленную анемией; лихорадка, вызванная инфекцией вследствие нейтропении; кровоточивость вследствие тромбоцитопении;
проявления, обусловленные ростом опухоли и неопластической инфильтрацией (чаще наблюдаются при ОЛЛ), включая боль в костях вследствие экспансии костного мозга и ин-
фильтрации поднадкостничногоЭпространства; генерализованная лимфаденопатия, сплено-
мегалия и гепатомегалия; увеличение тестикул; осложнения, связанные со сдавливанием крупных сосудов и дыхательных путей в средостении (при Т-ОЛЛ);
проявления со стороны ЦНС, например головная боль, рвота и парезы, возникающие в результате распространения процесса на оболочки головного мозга (эти проявления более характерны для ОЛЛ) /6/.
Периферические В-клеточные неоплазии
Хроническая лимфоцитарная лейкемия/мелкоклеточная лимфоцитарная лимфома. Эти два состоянияГразличаются только по степени лимфоцитоза в периферической крови. У большинства пациентов лимфоцитоз соответствует диагностическому критерию хронической лимфоцитарной лейкемии (ХЛЛ) (абсолютное количество лимфоцитов >4000 клеток/мм3). Средний возраст заболеваемости – 60 лет. Соотношение мужчин и женщин составляет 2:1. На долю мелкоклеточной лимфоцитарной лимфомы (МЛЛ) приходится только 4% неходжкинских лимфом /7/.
Лимфоузлы диффузно заполнены инфильтратом, состоящим преимущественно из малых лимфоцитов диаметром 6–12 мкм, с круглыми или слегка неправильной формы ядрами, конденсированным ядерным хроматином и скудной цитоплазмой (рис. 9.3). Также присутствует разное количество пролимфоцитов (крупных клеток с хорошо заметным одиночным, центрально расположенным ядром). Они часто образуют рыхлые агрегаты, называемые центрами пролиферации, которые содержат митотически активные клетки. Наличие центров пролиферации служит патогномоничным признаком ХЛЛ/МЛЛ. Кровь содержит большое количество малых круглых лимфоцитов с конденсированным ядерным хроматином и скудной цитоплазмой. Некоторые из этих клеток повреждаются в процессе приготовления мазков, образуя дегенерирующие лейкоциты (так называемые «тени Гумпрехта»). Костный мозг почти всегда содержит интерстициальные инфильтраты или агрегаты опухолевых клеток. Практически всегда инфильтраты обнаруживаются также в белой и красной пульпе селезенки и по ходу сосудов портальной системы печени /6/.
Костный мозг плоских и трубчатых костей красного цвета, но в отличие от миелоидного лейкоза в диафизах трубчатых костей среди красного костного мозга встречаются участки желтого цвета. В тяжелых случаях вся миелоидная ткань костного мозга диффузно вытесняется лейкозным лимфоцитарным инфильтратом, и сохранными остаются лишь небольшие островки миелоидного кроветворения.
166

БОЛЕЗНИ СИСТЕМЫ КРОВИ |
9 |
|
|
Рис. 9.3. ХЛЛ/МЛЛ (лимфоузел). А – при малом увеличенииЦвидно диффузное разрушение структуры лимфоузла; Б – при большом увеличении видно, что большинство опухолевых клеток представляют собой округлые малые лимфоциты. Также присутствует пролимфоцит – крупная клетка с центрально расположенным ядром (стрелка) /6/.
Отмечается генерализованное увеличение лимфатических узлов, которые сливаются в огромные мягкие или плотноватые пакеты. На разрезе они сочные, бело-розовые, однородные. Под микроско-
пом определяется утрата их архитектоники, вместо которой видны сплошные массы лимфоцитов. Граница между лимфатическимиЭузлами, как правило, сохранена, но нередко опухолевые лимфоци-
ты инфильтрируют капсулу лимфатических узлов и окружающие их ткани. В связи с лейкозной инфильтрацией увеличиваются размеры миндалин, лимфатических фолликулов кишечника, которые также представляют сочную бело-розовую ткань.
Селезенка достигает значительных размеров, масса ее увеличивается, она мясистой консистенции, красного цвета на разрезе. Лейкозный лимфоцитарный инфильтрат поражает, прежде всего, фолликулы, которые становятся крупными и сливаются. Затем лимфоциты разрастаются в красной пульпе, стенках сосудов, трабекулах и капсуле селезенки.
Печень увеличена, плотновата, на разрезе светло-коричневая. Нередко с поверхности и на разрезе видны мелкиеГсеро-белые узелки. Лимфоцитарная инфильтрация происходит главным образом по ходу волокнистой капсулы и портальных трактов. Гепатоциты находятся в состоянии белковой или жировой дистрофии.
Почки увеличены, плотноваты, серо-коричневого цвета. Лейкозная инфильтрация их бывает столь резко выражена, что структура почек на разрезе не выявляется. Лейкозная лимфоцитарная инфильтрация отмечается во многих органах и тканях (средостение, брыжейка, миокард, серозные и слизистые оболочки), она бывает диффузной и очаговой. Развиваются инфекционные осложнения (например, пневмонии) и гемолитические состояния (развитие гемолитической желтухи, общего гемосидероза), синдром сдавливания (например, сдавливание сердца, пищевода, трахеи при поражении лимфатических узлов средостения; сдавливание воротной вены и ее разветвлений с развитием портальной гипертензии и асцита при поражении лимфатических узлов брыжейки и ворот печени).
По иммунофенотипу этот вид лейкоза – новообразование из относительно зрелых В-лимфоцитов, экспрессирующих общие для В-клеток маркеры: СD19, СD20, СD23, SigIg, IgD, а также легкие цепи Ig. В 50% случаев обнаруживаются хромосомные аберрации, из них трисомия 12 – самая частая /5/.
Фолликулярная лимфома – наиболее часто встречающаяся форма вялотекущей неходжкинской лимфомы. Опухоль обычно возникает в среднем возрасте, с одинаковой частотой среди мужчин и женщин. Опухоль развивается из В-клеток герминативного центра и, вероятно, ассоциируется с хромосомной транслокацией гена BCL2.
167

ОБЩАЯ ПАТОЛОГИЧЕСКАЯ АНАТОМИЯ
Микроскопическое исследование пораженных лимфоузлов в большинстве случаев выявляет узелковую или узелковую и диффузную формы их роста разной пропорции присутствуют клетки двух основных типов (рис. 9.4):
небольшие клетки с ядрами неправильной или расщепленной формы и скудной цитоплаз-
мой (центроциты);
более крупные клетки с разреженным ядерным хроматином, несколькими ядрышками и умеренным количеством цитоплазмы (центробласты).
10% случаев в периферической крови присутствует лимфоцитоз. Костный мозг вовлекается в 85% случаев и характеризуется формированием паратрабекулярных лимфоидных агрегатов. В процесс часто вовлекаются также белая пульпа селезенки и портальные триады печени.
Во многих фолликулярных лимфомах малые расщепленныеЦклетки составляют большинство. В
Рис. 9.4. Фолликулярная лимфома (лимфоузел).
А – при малом увеличении видно, что лимфоузел запол- Энен узелковыми агрегатами клеток лимфомы;
Б – при большом увеличении видны малые лимфоидные клетки с конденсированным хроматином и ядрами неправильной или расщепленной формы (центроциты) и
более крупные клетки, содержащие несколько ядрышек
(центробласты) /6/.
ОбычноГфолликулярную лимфому описывают как безболезненную, генерализованную лимфаденопатию. Относительно нечасто в процесс вовлекаются экстранодальные участки: ЖКТ, ЦНС или яички. Трансформация фолликулярной лимфомы происходит в 30–50% случаев. Наиболее часто она трансформируется в диффузную В-крупноклеточную лимфому, реже возникает опухоль, напоминающая лимфому Беркитта и ассоциированная с транслокацией c-MYC. Как и в В-клетках герминативного центра, в клетках фолликулярной лимфомы постоянно происходят соматические гипермутации, которые способствуют трансформации за счет точечных мутаций или хромосомных аберраций.
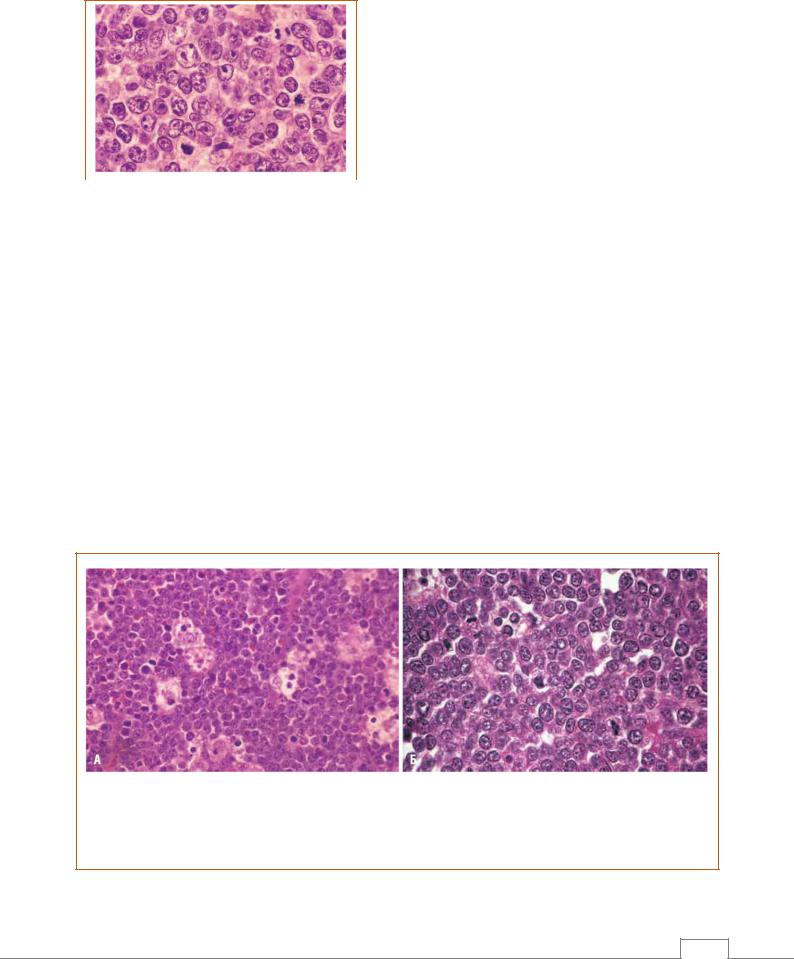
Диффузная В-крупноклеточная лимфома – наиболее часто встречающаяся форма неходжкинских лимфом. Опухоль чаще возникает у мужчин. Средний возраст пациента составляет ~60 лет, однако диффузная В-крупноклеточная лимфома встречается также у молодых людей и детей.
Общими признаками служат относительно большой размер опухолевых клеток (диаметр обычно в 4–5 раз превышает диаметр малого лимфоцита) и диффузный тип роста (рис. 9.5), однако возможны значительные морфологические вариации. Опухолевые клетки чаще имеют круглое или овальное ядро, которое выглядит как пузырек вследствие скопления хроматина у ядерной мембраны. В некоторых случаях ядра могут быть многодольча-тыми или расщепленными. Клетки могут иметь 2–3 ядрышка, прилегающих к ядерной мембране, или одно центрально расположенное ядрышко. Количество цитоплазмы обычно умеренное, она бледная или базофильная. Более анапластические опухоли могут содержать даже многоядерные клетки с крупными, похожими на включения ядрышками. Эти клетки напоминают злокачественные клетки лимфомы Ходжкина (клетки Рид-Штернберга).
168
БОЛЕЗНИ СИСТЕМЫ КРОВИ |
9 |
|
|
Рис. 9.5. Диффузная В-крупноклеточная лимфома.
Опухолевые клетки содержат крупные ядра, рыхлый хроматин и хорошо заметные ядрышки /6/.
|
|
|
|
|
Ц |
|
|
|
Диффузная |
|
|
- |
|
|
|
|
|
|
||
невой |
|
|
|
|
любом |
|
участке |
|
|
|
|
|
|
|
|
аденоиды |
|
|
|
большой |
|
|
деструктивной |
|
|
|
- |
же, костях |
|
|
|
|
||
ходе |
|
Э |
|
|
||
|
|
Лимфома |
|
|
||
|
|
|
|
|
|
|
|
|
эндемическая (африканская) лимфома Беркитта; |
|
|
||
|
|
спорадическая (неафриканская) лимфома Беркитта; |
|
|
||
|
|
лимфома Беркитта, ассоциированная с иммунодефицитом (чаще всего наблюдается у лиц с |
||||
|
|
ВИЧ-инфекцией). |
|
|
|
|
|
|
Все |
|
|
|
- |
скими |
|
|
|
|
|
|
|
|
Пораженная |
|
|
|
клеток |
|
|
промежуточной |
|
|
|
|
ядерным |
|
|
|
|
|
|
Рис. 9.6. Лимфома Беркитта. А – при малом увеличении видны многочисленные бледные макрофаги, содержащие окрашивающиеся включения, создающие картину «звездного неба»; Б – при большом увеличении в опухолевых клетках можно видеть многочисленные мелкие ядрышки, характерен высокий митотический индекс. Отсутствие существенных вариаций формы и величины ядер создает однообразную картину /6/.
Опухоль имеет высокий митотический индекс и содержит многочисленные апоптотические клетки, остатки ядер которых фагоцитируются рассеянными нормальными макрофагами. Эти фаго-
169

ОБЩАЯ ПАТОЛОГИЧЕСКАЯ АНАТОМИЯ
циты содержат большое количество прозрачной цитоплазмы, создавая характерную картину «звездного неба» (starry sky). При вовлечении костного мозга в аспиратах обнаруживаются опухолевые клетки с умеренно глыбчатым ядерным хроматином, 2–5 различимыми ядрышками и ярко-синей цитоплазмой, содержащей прозрачные цитоплазматические вакуоли.
Эндемические и спорадические лимфомы Беркитта наблюдаются главным образом у детей и молодых взрослых. Большинство опухолей манифестирует в экстранодальных участках. Эндемическая лимфома Беркитта часто локализуется на нижней челюсти, также характерна необычная локализа-
гаммапатия», «диспротеинемия» и «парапротеинемия». С Цмоноклональ-ными гаммапатиями ассоциируются следующие клинико-патологические формы:
ция – в органах брюшной полости, особенно в почках, яичниках и надпочечниках. Спорадическая лимфома Беркитта обычно представляет собой опухолевую массу в илеоцекальной области и брюшине. Поражение костного мозга и периферической крови встречается редко, особенно в эндемических случаях. Лимфома Беркитта очень агрессивна, однако хорошо поддается химиотерапии.
Плазмоклеточные неоплазии и родственные опухоли. Эти В-клеточные опухоли содержат неопластические плазматические клетки, практически всегда секретирующие моноклональный Ig или фрагмент Ig. Все плазмоклеточные неоплазии (называемые также дискразиями) обусловливают >15% летальных исходов, вызываемых лимфоидными неоплазиями. Наиболее частая и опасная из
этих неоплазий – множественная миелома.
Моноклональные антитела, определяемые в крови, получили название «М-белок». Поскольку полный М-белок имеет молекулярную массу 160 кДа или более, его присутствие в организме ограничено плазмой и внеклеточной жидкостью и в отсутствие поражений почечных клубочков он не попадает в мочу. Однако в отличие от нормальных плазматических клеток, в которых продукция Ig и соединение легких и тяжелых цепей Ig тесно связаны, неопластические плазматические клетки часто синтезируют избыток легких или тяжелых цепей вместе с полными Ig. Иногда образуются только легкие или тяжелые цепи Ig. Свободные легкие цепи Ig (белок Бенс-Джонса) достаточно малы и экс-
кретируются с мочой.
Для описания продукции аномальных Ig используют специальные термины: «моноклональная
появлению симптомов, обусловленныхЭповышенной вязкостью крови. Синдром наблюдается у пожилых, чаще всего в сочетании с лимфоплазмоцитарной лимфомой;
Гболезнь тяжелых цепей (средиземноморская лимфома) – редкая моноклональная гаммапатия, ко-
множественная миелома – наиболее важная моноклональная гаммапатия, которая представляет собой опухолевые массы из плазматических клеток, рассеянных в тканях костей скелета. Солитарная плазмоцитома – редкий вариант опухоли из плазматических клеток, характеризующийся одиночным новообразованием в костях или мягких тканях. Термин «тлеющая миелома» относится к другому редкому варианту с отсутствием симптомов и высоким содержанием
М-белка в плазме;
макроглобулинемия Вальденстрема – синдром, при котором высокий уровень IgM приводит к
торая сопровождается разнообразными расстройствами, включая лимфоплазмоцитарную лимфому и лимфому из клеток маргинальной зоны необычной локализации в тонкой кишке, возникающую в популяциях с дефицитом питания. Общие признаки – синтез и секреция свободных фрагментов тяжелых цепей;
первичный, или ассоциированный с иммуноцитами, амилоидов – клинико-патологическая форма,
возникающая в результате моноклональной пролиферации плазматических клеток, секретирующих легкие цепи (обычно изотипа А), откладывающиеся в виде амилоида. Некоторые пациенты имеют явную множественную миелому, тогда как у других в костном мозге присутствует лишь незначительная клональная популяция плазматических клеток;
170

БОЛЕЗНИ СИСТЕМЫ КРОВИ |
9 |
|
|
моноклональная гаммапатия неустановленной значимости – клинико-патологическая форма без симптомов, но с присутствием в крови малого или умеренно большого М-белка. Моноклональная гаммапатия неустановленной значимости очень часто встречается в пожилом возрасте. Отмечается стабильно низкая частота ее трансформации в симптоматическую моноклональную гаммапатию, чаще в множественную миелому.
Множественная миелома – самое частое заболевание из группы плазмоклеточных дискразий.
Миелома характеризуется клональной пролиферацией опухолевых плазматических клеток в костном
мозге, что приводит к появлению в костях скелета множественныхЦочагов лизиса, именно поражение
костного мозга дало название болезни. При этом заболевании происходит пролиферация одного клеточного клона, вырабатывающего моноклональный Ig с одной тяжелой цепью, одним типом легкой цепи и идентичными последовательностями аминокислотных остатков в переменных районах каждой цепи. Эти моноклональные белки определяются в крови и являются парапротеинами, или миеломными белками. Моноклональные иммуноглобулины могут иметь любой изотип, хотя самым частым (у 50% больных) является IgG. У некоторых больных (у 20%) синтезируются только легкие цепи – ϰ или λ. Благодаря низкой молекулярной массе (25 кДа) легкие цепи в качестве мономеров или димеров выделяются с мочой и выпадают в осадок при охлаждении. Они известны под названием белка Бенс-Джонса (криоглобулина). У таких больных в моче имеется белок Бенс-Джонса, но в плазме нет М-компонента (болезнь легких цепей). У 80% пациентов малигнизированные плазматические клетки синтезируют как полныйЭIg, так и избыток легких цепей, у них можно выявить как белок БенеДжонса, так и М-компонент.
При множественной миеломе пролиферация опухолевых плазматических клеток, как правило, ограничена пределами костного мозга, но может распространяться и на другие ткани. Болезнь встречается у пожилых лиц – средний возраст 60 лет. Диагностика основывается на выявлении остеолитических поражений при рентгеноскопии скелета, моноклонального белка в сыворотке крови или моче, а также типичных клеточных изменений в костном мозге.
Этиология и патогенез неизвестны. Пролиферация миеломных клеток поддерживается цитокином IL-6, который вырабатывают фибробласты и макрофаги стромы костного мозга. Во многих случаях приГмиеломе выявлена хромосомная транслокация в области локуса IgG на хромосоме 14. Хромосомные аберрации в 4-й хромосоме могут приводить к изменениям гена, кодирующего рецептор к фактору роста фибробластов 3, в результате мутации рецептор сохраняет постоянную активность.
Главными патоморфологическими признаками являются деструктивные изменения в скелете. На рентгенограммах и макроскопических препаратах они напоминают выбитые отверстия (или пробоины). В процесс может быть вовлечена любая кость, но чаще поражаются позвоночник, ребра, кости черепа, реже – кости таза, бедренная кость, ключица и лопатка. Часто отмечаются вертебральный коллапс (острая деформация позвоночника) и патологические переломы костей. Опухолевая ткань растет из полостей костного мозга по направлению к корковой зоне кости. Резорбция костного вещества обусловлена секрецией факторов, активирующих остеокласты. Иногда выраженное распространение опухолевой ткани внутри костей приводит к диффузному остеопорозу /3/.
По характеру опухолевых инфильтратов, развивающиеся в костном мозге и в костях выделяются нижеследующие морфологические формы:
узелковая (очаговая) форма. Обычно развивается на ранних стадиях заболевания;
диффузная форма. Сопровождается прогрессированием заболевания. Миеломные клетки, увеличиваясь с высокой скоростью, распрастраняются в диффузном виде в костном мозге. При этом, нарушается процесс нормального гемопоэза, со стороны миеломных клеток стромы костного мозга – ретикулиновые волокна разрушаются, опухолевая инфильтрация входит в костную ткань и становится причиной ее деструкции – остеолизиса и остеопороза;
171

ОБЩАЯ ПАТОЛОГИЧЕСКАЯ АНАТОМИЯ
диффузно-узелковая форма. При этом, в костном мозге, видны очаговые инфильтраты миеломных клеток на фоне инфильтрации диффузных лейкозных клеток /1/.
Экстрамедуллярные опухолевые пролифераты наблюдаются в селезенке, печени и лимфатических узлах. На поздних стадиях болезни поражаются и другие органы, очень редко развивается плазмоцитарный лейкоз.
В цитологических мазках многие клетки выглядят как нормальные зрелые плазмоциты, но часто преобладают более примитивные элементы – плазмобласты и клетки, имеющие промежуточное
строение между лимфоцитом и плазматической клеткой.ЦНередко определяются 2- и 3-ядерные
клетки, а также оксифильные внутриклеточные белковые агрегаты, известные под названием телец Русселя. Надо отметить, что плазматические клетки имеются и в нормальном костном мозге, их количество может возрастать при хронических воспалительных заболеваниях, но оно редко превышает 10% всех клеток костного мозга.
Замещение костного мозга опухолевой тканью приводит к нормохромной нормоцитарной ане-
мии, нейтропении и тромбоцитопении, что повышает чувствительность к инфекциям, которые чаще
всего становятся причиной смерти. Развивается синдром повышенной вязкости крови, сходный с та-
ковым при макроглобулинемии Вальденстрема. Парапротеин вызывает формирование «монетных
столбиков», т.е. неустойчивых агрегатов эритроцитов, сладж эритроцитов (более устойчивая их агре-
гация), а также обусловливает высокую СОЭ. При миеломе легких цепей СОЭ не повышается. У 30% больных встречается гиперкальциемияЭ, вызванная интенсивной резорбцией костного вещества /3/.
С клинической точки зрения, внутри парапротеинемических патологий, большое значение имеют изменения в почках (миеломная нефропатия или парапротеинемический нефроз). При этом, в почечной ткане, в мезангиальных областях клубочков накапливаются белки Бенс-Джонса, корковые и мозговые вещества почки постепенно склерозируются, а также развивается вторичный ALамилоидоз, что приводит к смерти больных /1/.
Макроглобулинемия Вальденстрема развивается преимущественно у мужчин старше 50 лет, составляет около 5% всех плазмоцитарных заболеваний, построена из В-клеток разного вида – от мелких округлых лимфоцитов до плазматических клеток. Определяется М-компонент, в основном связанныйГс моноклональной продукцией IgМ.
Клинически опухоль напоминает другие лимфомы, опухолевые В-лимфоциты инфильтрируют лимфатические узлы, костный мозг и селезенку. Резорбция костей не наблюдается. Характерный признак болезни – продукция больших количеств IgМ, его содержание в сыворотке крови составляет 25–80 г/л. Клиническая картина обусловлена повышенной вязкостью крови, появляется неврологическая симптоматика с головокружениями, парезами и ухудшением зрения (ретинопатия). Встречаются небольшие кровоизлияния в слизистые оболочки вследствие либо нарушения кровотока, либо снижения функции тромбоцитов (геморрагический синдром).
Болезни тяжелых цепей (болезнь Франклина) представляют В-клеточные опухоли с гетероген-
ной клинической и морфологической картиной и секрецией тяжелых цепей (Н-цепи) различных классов Ig. Лимфоидная пролиферация в тканях сопровождается ведущим признаком – наличием в крови или моче специфических тяжелых цепей Ig. В соответствии с классом тяжелых цепей (γ, δ, α, μ) выделяют несколько вариантов заболевания. Болезни тяжелых цепей характеризуются плохим прогнозом /3/.
Лимфома из клеток мантийной зоны – редкая лимфоидная неоплазия. Она обычно появляется в возрасте 40–60 лет преимущественно у лиц мужского пола. Клетки этой лимфомы очень похожи на нормальные В-клетки мантийной зоны, окружающей герминативные центры /7/.
Опухолевые клетки в лимфоузлах могут окружать реактивные герминативные центры, придавая им узелковый вид (при малом увеличении) или диффузно заполняя узел. Как правило, пролиферирующий компонент состоит из гомогенной популяции малых лимфоцитов с ядрами неправильной
172