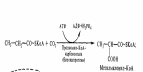№25
.docxНаследственные нарушения обмена аминокислот
Нарушение |
Дефектная реакция (формулами) |
Значение реакции (и метаболического пути, стадией которого является данная реакция |
Дефектный фермент/белок |
Биохимический профиль |
Клинические проявления |
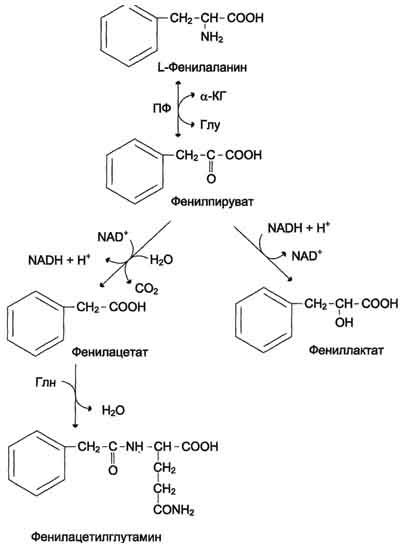
1.Фенилкетонурия, |
|
В печени здоровых людей небольшая часть фенилаланина (∼10%) превращается в фенил-лактат и фенилацетилглутамин. Этот путь катаболизма фенилаланина становится главным при нарушении основного пути - превращения в тирозин, катализируемого фенил-аланингидроксилазой. Такое нарушение сопровождается гиперфенилаланинемией и повышением в крови и моче содержания метаболитов альтернативного пути: фенилпирувата, фенилацетата, фениллактата и фенилацетилглутамина. |
Фенилаланингидроксилаза |
Концентрация фенилаланина повышается в крови в 20-30 раз (в норме - 1,0-2,0 мг/дл), в моче - в 100-300 раз по сравнению с нормой (30 мг/дл). Концентрация фенилпирувата и фениллактата в моче достигает 300-600 мг/дл при полном отсутствии в норме |
Проявления ФКУ - нарушение умственного и физического развития, судорожный синдром, нарушение пигментации. При отсутствии лечения больные не доживают до 30 лет. Частота заболевания - 1:10 000 новорождённых. Заболевание наследуется по аутосомно-рецессивному типу. Тяжёлые проявления ФКУ связаны с токсическим действием на клетки мозга высоких концентраций фенилаланина, фенилпирувата, фениллактата. Большие концентрации фенилаланина ограничивают транспорт тирозина и триптофана через гематоэнцефаличеекий барьер и тормозят синтез нейромедиаторов (дофамина, норадреналина, серотонина). |
2.Гомоцистинурия |
|
Данная реация часть метаболического пути метионина,который в свою очередь необходим для синтеза белков, синтеза цистеина, Явл донором метильных групп для синтеза многих соединений в реакции трансметилирования |
Цистатионинсинтаза; Дефицит B6 |
При гомоцистинурии в моче, плазме крови, ликворе обнаруживаются значительные количества гомоцистина, повышение содержания метионина при сниженном уровне цистина. В биоптатах и печени выявляется специфический ферментативный дефект. |
Гомоцистинурия сопровождается умственной отсталостью, судорожным синдромом, подвывихом хрусталиков, катарактой, глаукомой, атрофией зрительных нервов, деформацией грудной клетки, сколиозом, арахнодактилией, артериальными и венозными тромбозами. |
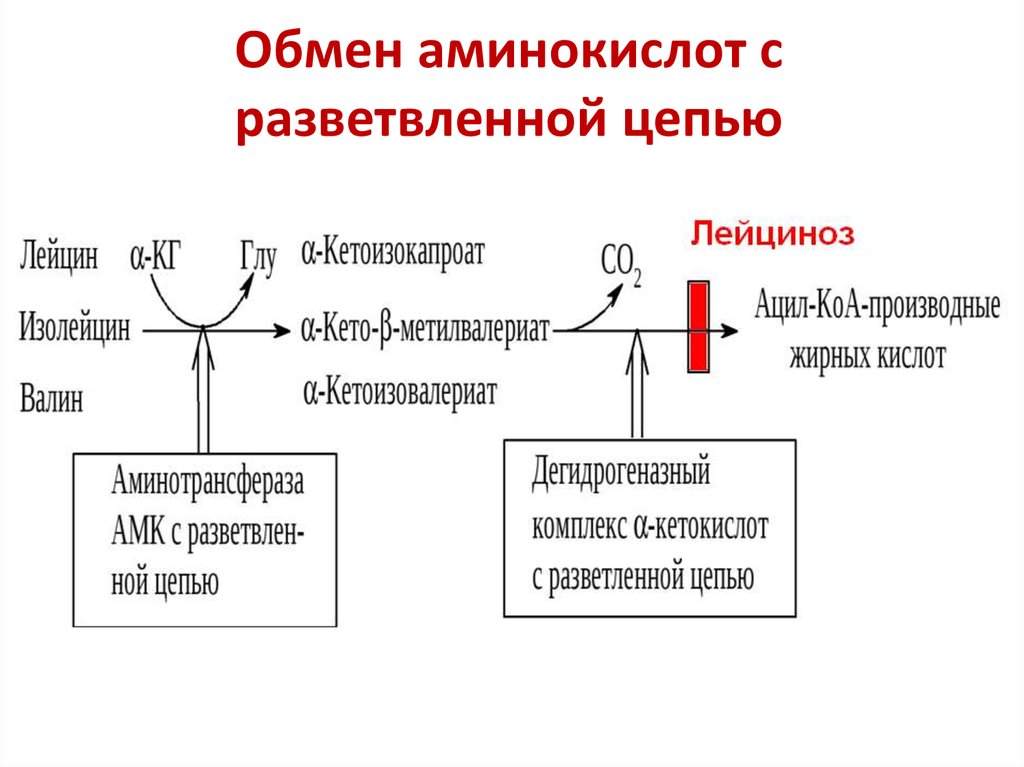
3.Лейциноз. |
|
Обмен Аминокислот с разветвленной цепью(Лейцина, Валина, Изолейцина) |
Дегидрогеназный комплекс альфа-кетокислот с развлетвленной цепью (фермент декарбоксилазы альфа-кетокислот) |
Снижение или полное отсутствие активности фермента декарбоксилазы альфа-кетокислот, что приводит к накоплению в организме Лейцина |
Ребенок отказывается от кормления и монотонное плачет. При попытках накормить возникает рвота и срыгивание. Специфический запах мочи - кленового сиропа. |
4.Болезнь "'кленового сиропа" |
|
При распаде валина, лейцина, изолейцина, они проходят ряд этапов: трансаминирование с получением соответствующих α-кетокислот, их окислительное декарбоксилирование, еще одно окисление с образованием ненасыщенных кетокислот и уже более индивидуальные реакции превращения. Конечными продуктами распада являются для лейцина только ацетил-SКоА, для изолейцина и валина - ацетил-SКоА и сукцинил-SКоА. Нарушается метаболизм аминокислот лейцина, изолейцина, валина. Патогенез заболевания связан с нарушением обмена и Дефицит дегидрогеназы кетокислот с разветвленной цепью В плазме крови, в пятнах высушенной крови выявляют повышение концентрации лейцина, изолейцина и валина, а так же 2- кетоизокапроновой, 2-кето-3-метилвалериановой, 2-кетоизовалериановой кислот. Заболевание проявляется с первой недели жизни с внезапного ухудшения состояния после кормления грудным молоком или молочной смесью. Ребенок вял, отказывается от еды, появляется рвота, возможны судороги, потеря сознания, цианоз, нарушение дыхания, коматозное состояние. Характерны потеря веса и прогрессирующая неврологическая симптоматика в виде нарушения мышечного тонуса, патологического рефлекса Моро. Дылдина накоплением аминокислот, накоплением в биологических жидкостях продуктов обмена – 2- кетоизокапроновой, 2-кето-3-метилвалериановой, 2-кетоизовалериановой кислот |
Дефицит дегидрогеназы кетокислот с разветвленной цепью |
В плазме крови, в пятнах высушенной крови выявляют повышение концентрации лейцина, изолейцина и валина, а так же 2- кетоизокапроновой, 2-кето-3-метилвалериановой, 2-кетоизовалериановой кислот. |
Заболевание проявляется с первой недели жизни с внезапного ухудшения состояния после кормления грудным молоком или молочной смесью. Ребенок вял, отказывается от еды, появляется рвота, возможны судороги, потеря сознания, цианоз, нарушение дыхания, коматозное состояние. Характерны потеря веса и прогрессирующая неврологическая симптоматика в виде нарушения мышечного тонуса, патологического рефлекса Моро |
5.Изовалериановая ацидемия |
|
изовалерил-КоА дегидрогеназу, участвует в обмене лейцина и переводит изовалерил-КоА в 3-метилкротонил-КоА.( бетта- метилкротонил- skoa) |
при данной патологии происходит дефект изовалерил-KoA дегидрогеназы |
основан на выявлении повышенного уровня изовалериановой кислоты и ее метаболитов в моче, а в крови изовалерилкарнитина |
большинстве случаев заболевание имеет кризовое течение. Метаболический криз провоцируется факторами, ведущими к усилению процессов катаболизма: интеркуррентные инфекции, хирургическое вмешательство, прием больших количеств белка и др. Острая неонатальная форма характеризуется манифестацией на первой неделе жизни: упорная рвота, отказ от еды, снижение массы тела, резкая вялость, сонливость, летаргия, угнетение ЦНС, судороги. Характерен необычный запах мочи типа “потных ног” или “сыра”. Заболевание протекает тяжело и у 30% детей приводит к летальному исходу. |
6.Пропионовая ацидемия |
|
Метилмалонил-КоA — связанная с коферментом А форма метилмалоновой кислоты, преобразуется в сукцинил-КоА посредством метилмалонил-КоА мутазы, в реакции, что требует витамина B12 в качестве кофактора. Таким образом, она входит в цикл Кребса, и является одной из составляющих заживляющих реакций. |
Пропионил-СоА карбоксилаза |
Повышенный уровень метаболитов пропионовой кислоты, в том числе и тиглита метилцитрата и их глициновых конъюгатов в крови и моче, а также происходит измерение активности пропионил-СоА-карбоксилазы в лейкоцитах или культивируемых фибробластах и/или генетических тестах |
Плохой аппетит, рвота и расстройства дыхания из-за глубокого метаболического ацидоза с дефицитом анионов, гипогликемия и гипераммониемия. Могут возникать судороги, угнетение костного мозга является распространенным явлением. У выживших могут развиваться трубчатая нефропатия, умственная отсталость и неврологические нарушения. |
7.Метилмалоновая ацидемия |
|
Окисление жирных кислот с нечетным числом С-атомов. Суть превращения Пропионил-SkoA сводится к его карбоксилированию, изомеризщации и образованию сукцинил-SkoA |
Метилмалонил-КоА-мутаза |
Диагноз ставится на основе анализа уровня аминокислот изолейцина, валина, метионина и треонина в крови, почечной экскреции органических кислот - метилмалоновой, 3-гидроксипропионовой, 3-гидрокси-n-валериановой, метиллимонной, пропионилглицина |
Первые симптомы обычно появляются в возрасте 2 недель – 4 месяцев: упорная рвота, отказ от еды, дегидратация, вялость, сонливость, дыхательные нарушения, задержка психомоторного и физического развития, иногда развиваются инсультоподобные эпизоды. В более старшем возрасте, помимо значительной задержки психоречевого и моторного развития, у детей отмечаются неврологические нарушения, в виде различных эксрапирамидных нарушений (хореоатетоидные и миоклонические гиперкинезы, мышечная дистония, инсультоподобные эпизоды, эпилептические приступы), поражение почек по типу тубулоинтерстициального нефрита с артериальной гипертензией и почечной недостаточностью, эритематозный дерматит, в отдельных случаях - панкреатит и кардиомиопатия. |
8.Дефицит сульфит оксидазы |
|
Сульфит оксидаза превращает сульфит в сульфат на последнем этапе деградации цистеина и метионина; ферменту требуется молибденовый кофактор. |
Сульфит оксидаза/ метионин, цистеин |
Диагноз ставится на основе повышенного уровня сульфита в моче и подтверждается измерением уровней ферментов в фибробластах и уровня кофактора в образцах печени при биопсии и генетических тестах. |
Заболевание вызывает неврологические расстройства, умственные и физические отклонения, деградацию мозга и смерть. |
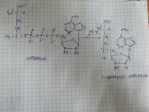
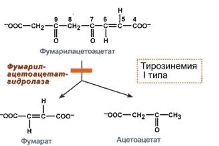
9.Тирозинемия типа I |
|
Фумарат используется для синтеза глюкозы или окисляется до CO2 и H2O. Ацелоацетат окисляется в тканях с выделением энергии.
|
Недостаточностью фермента фумарилацетоацетат-гидролазы. |
Накапливается фумарилацетоацетат и его метаболиты (сукцинилацетон) в крови. |
Острая форма: тирозинемии с началом в возрасте 2-7 мес и смертью 90% больных в возрасте 1-2 года из-за недостаточности печени. Симптомы гипотрофия, рвота, "капустный запах" от тела и мочи, задержка развития, кровоточивость, диарея, мелена, гематурия, желтуха, анемия, периферические невропатии и параличи, кардиомиопатия, слабость мышц, дыхательные нарушения, гипогликемия. Хроническая форма: болезнь развивается позднее, медленнее прогрессирует. Продолжительность жизни около 10 лет. Наблюдаются гипотрофия, узелковый цирроз печени и печеночная недостаточность, множественные дефекты почечной канальцевой реабсорбции с появлением синдрома Фанкони (щелочная рН мочи, глюкозурия, протеинурия), аминоацидурия, лейкопения, тромбоцитопения, рахитоподобные заболевания (остеопороз, остеомаляция), симптомы, напоминающие острую порфирию, умственная отсталость и неврологические изменения.
|
10.Тирозинемия типа II и Тирозинемия Тип III |
- |
- |
- |
- |
- |
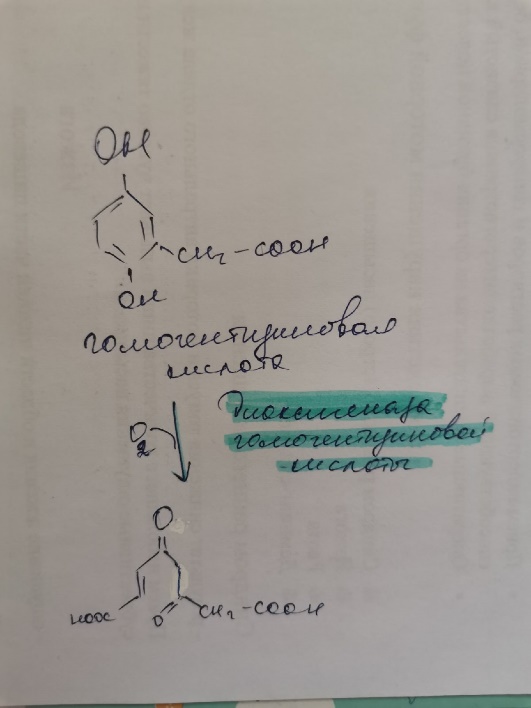
11.Алкаптонурия |
|
Диоксигеназа гомогентизиновой кислоты участвует в обмене тирозина и переводит гомогетензиновую кислоту в фумарилацетоацетат, а также является важной частью превращения тирозина |
Дефект диоксигеназы гомогентизиновой кислоты |
В моче высокие концентрации гомогентизиновой кислоты |
Потемнение мочи на воздухе, пигментация соединительной ткани (охроноз) и артрит |
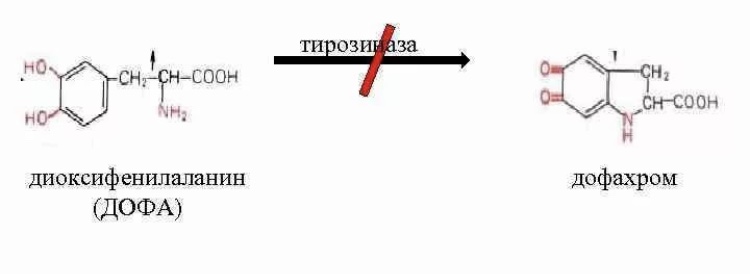
12. Альбинизм |
|
Причиной альбинизма как правило, является метаболический дефект, связанный с потерей меланоцитами способности к синтезу меланина вследствие отсутствия фермента тирозиназы, который окисляет диоксифенилаланин в дофахром, являющийся предшественником фермента меланина |
Тирозиназа |
без изменений |
фенотипически проявляющийся отсутствием присущей для данного вида окраски кожи, волос, шерсти, радужной и пигментной оболочек глаз, зелёных частей растений.
|
13. Некетоновая гиперглицинемия |
|
Глицин является предшественником порфирина, пуриновых оснований, глутатиона. |
Р-протеин (ПФДК) Т-протеин (АМТ) Н-протеин (белок-переносчик ВСРГ) L-протеин (липоамид дегидрогеназа) |
повышение уровня глицина в: крови (в 10–20 раз), моче и ликворе (в 10–30 раз) при отсутствии ацидоза, кетоза, органической ацидурии |
Вялость, мышечная гипотония, сниженная двигательная активность, выраженная сонливость (вплоть до литургии), отказ от еды, миоклонические судорожные приступы, опистотонус. Тяжелая эпилептическая энцефалопатия, спастический церебральный паралич, гипоплазия мозолистого тела. |
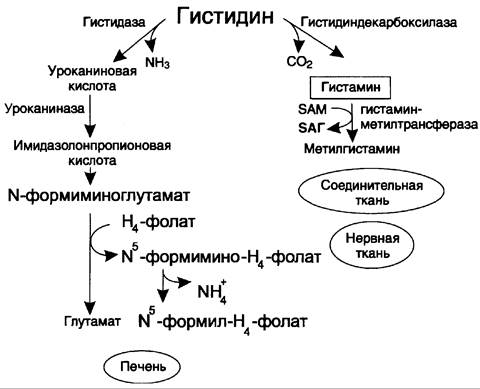
14.Гистидинемия |
Левая часть схемы внизу, реакция – первая
|
Значение реакции – образование уроканиновой кислоты, выделение аммиака(токсичность, влияние на цнс), Значение пути: образование глутамата ( предшестенник ГАМК-торм.медиатора) и гистамина. Основные функции гистамина 1.Защитная: как медиатор воспаления защищает организм от воздействия агрессивных факторов внешней среды. 2. Является пищеварительным гормоном: стимулирует выделение слюны и желудочного сока 3.Является нейромедиатором, ответственным за регуляцию сна и бодрствования: активирует нервные клетки, противодействует сну.
|
Гистидаза |
Увеличение гистидина в крови и других биологических жидкостях, в крови концентрация от 20 до 250 мг/л, при норме в 3-10 раз меньше; Повышается экскреция имидазолпировиноградной, имидазолмолочной, имидазолуксусной кислот; Отсутствие или низкая концентрация уроканиновой кислоты в поте и моче после нагрузки гистидином, В крови и моче наблюдается повышение концентрации аланина, в плазме – низкий уровень серотонина, в ликворе – снижение концентрации глютамина и глютаминовой кислоты, у некоторых больных выявляется повышение в крови уровня серина, треонина, глицина, агринина, орнитина |
Олигофрения, судороги, расстройства координации движений, нарушения речи (большая вариабельность – от тяжелой умственной отсталости и выраженных неврологической симптоматики до полного отсутствия симптомов) |
15. Болезнь Хартнупа |
|
Триптофан является главным субстратом для синтеза витамина РР, участвующий в тканевом дыхании и энергетических процесах. Также триптофан необходим для синтеза серотонина и мелатонина (гормона сна) |
Дефект триптофандиоксигеназы или Натрий-зависимого белка переносчика нейтральных аминокислот (локализованных в кишечнике и почечных канальца) |
1)В моче высокие концентрации нейтральных аминокислот (триптофана, тирозина, гистидина) 2) обнаружение в моче индола и индикана ( как продукта распада этих аминокислот) 3) в крови снижается концентрация триптофана 4)недостаточность витамина РР |
Неврологические, психические и дерматовенерологические проявление пеллагры, фоточувствительная кожная сыпь, эмоциональная лабильность, возможны энцефалопатия, преходящая мозжечковая атаксия, поражение печени и ЖКТ. симптом голубых пеленок |