

Патофизиология_дыхания,_ФОМ,_рус_,_2021г_
.pdfКафедра Патофизиологии, Патофизиология дыхания, ФОМ, 2021г.
•легочная эмболия,
•сердечная недостаточность и ишемическая болезнь сердца,
•анемии,
•тяжелая физическая работа,
•психоэмоциональный стресс и т.д.
Одышка имеет различные, часто сочетающиеся механизмы развития, обусловленные конкретной клинической ситуацией. Тем не менее в любом случае одышка является следствием гиперактивности бульбарного дыхательного центра или патологической гиперстимуляцией, обусловленной поступлением афферентных импульсов от различных структур (рис. 21.5):
•от ирритантных и J-рецепторов дыхательной системы,
•поступление сигнала по соматическим нервам от дыхательных мышц, других мышц, суставов,
•от хеморецепторов дыхательного центра, аорты и каротидных хеморецепторов,
•от центров коры головного мозга.
При одышке обычно наблюдается частое глубокое дыхание, хотя возможны другие комбинации глубины и частоты дыхания. Усиление и вдоха, и выдоха имеет активный характер, осуществляется посредством мышц, участвующих при выдохе. Различают инспираторную и экспираторную одышку. Инспираторная одышка характеризуется интенсивностью,
продолжительностью и затруднением вдоха.
Экспираторная одышка характеризуется затруднением выдоха.
Инспираторная одышка развивается при нарушении проходимости дыхательных путей (отек горла, частичная непроходимость рубцом, отеком, инородным телом). В этих случаях происходит медленное прохождение воздуха к альвеолам, повышается порог чувствительности механорецепторов легких.
Задерживается передача информации по блуждающему нерву (рефлекс Херринга-Брейера), вследствие чего увеличивается продолжительность фазы вдоха. Частыми причинами инспираторной одышки являются также патологические процессы (гидроторакс,
пневмоторакс, деформация грудной клетки) сдавливающие легкие и ограничивающие их движения.
Экспираторная одышка наблюдается при приступах бронхиальной астмы, обструктивных поражениях нижних дыхательных путей, в последствии увеличивается остаточный дыхательный объем, что вызывает возбуждение механорецепторов. В результате ускоряется передача информации по блуждающим нервам,
преждевременное формирование тормозного рефлекса Херринга-Брейера тормозит фазу вдоха и продлевает фазу выдоха.
11

Кафедра Патофизиологии, Патофизиология системы дыхания, ФОМ, 2021г.
21.3. ОСТРЫЙ ДЫХАТЕЛЬНЫЙ ДИСТРЕСС СИНДРОМ
Острый дыхательный дистресс синдром (ОРДС) называется также дыхательным дистресс синдромом взрослых, синдромом «влажного легкого», «шокового легкого» и т.д. Каждый год этот синдром развивается у
150000 - 200000 человек, 50-60% из которых умирают.
Во время вьетнамской войны американские врачи обратили внимание на то, что часто, спустя 2-3 дня после выведения больных из тяжелого травматического шока развивается специфический синдром дыхательной недостаточности. Впоследствие многочисленные клинические исследования были посвящены изучению этого феномена. В 1967г. Ashbaugh и соавторы это состояние назвали острым дыхательным дистресс синдромом взрослых, учитывая клинико-морфологическое сходство с респираторным дистресс синдромом новорожденных. Позже он был переименован в острый респираторный дистресс-синдром,
поскольку он может развиваться как у взрослых, так и у детей.
В середине 70-х годов прошлого века прошла новая волна ярых дискуссий, касающихся названия синдрома, и учитывая его многофакторность не считали нужным применение названия этого синдрома в медицине. И наконец, американско-европейской согласительной конференцией был принят не только термин, но были даны определение и критерии его диагностики.
ОРДС не является специфичным заболеванием, он рассматривается как синдром, характеризующийся воспалением и повышением проницаемости альвеоло-капиллярной мембраны, сочетающийся совокупностью клинических, рентгенологических и физиологических нарушений, не обусловленных гипертензией левого предсердия или легочной капиллярной гипертензией, которые однако могут наблюдаться одновременно
(определение, принятое согласительной американско-европейской конференцией, 1994 г.).
Мы отдаем предпочтение следующему определению. Острый респираторный дистресс-синдром - это тяжелая форма недостаточности дыхания, развивающаяся при прямом и/или системном повреждении альвеоло-капиллярной мембраны и характеризующаяся диффузной инфильтрацией легочной ткани и выраженной гипоксемией.
В2012 г. на Берлинской конференции были пересмотренны критерии ОРДС:
1.острое начало (признаки дыхательной недостаточности должны проявляться в течении одной недели в условиях уже существующей патологии, либо у больного должны проявляться новые клинические признаки или усугубление уже имеющихся в течении недели),
2.нарушение оксигенации, определяемое по дыхательному индексу: PaO2/FiO2<300 мм рт.ст. (где PaO2
Рис. 21.6. На рентгенограмме пациента видны диффузные, двухсторонние альвеолярные инфильтраты. Кардиомегалия отсутствует.
– парциальное давление кислорода в артериальной крови,
а FiO2 – концентрация кислорода во вдыхаемом воздухе в десятичном выражении, например 50%-ая концентрация соответствует приблизительно FiO2=0,5).
3. На рентгенограмме или КТ грудной клетки -
двухсторонняя инфильтрация легких (которая полностью не обьясняется наличием иной легочной патологии) (рис.
21.6).
4. У больного дыхательная недостаточность не должна полностью объясняться наличием сердечной недостаточности или перегрузкой жидкостью.
Объективное исследование (например, эхокардиография)
необходимо для исключения легочной гипертензии.
Этиология. ОРДС в принципе может развиться у
12

Кафедра Патофизиологии, Патофизиология дыхания, ФОМ, 2021г.
больного с любой патологией, если она сопровождается острым системным воспалением. В основе развития этого синдрома лежит отек легких, основные причины которого приведены на рисунке 21.7.
К развитию ОРДС приводят факторы, указанные в пункте Б. При этом сепсис, шок пневмонии,
аспирация жидкостью обуславливают развитие большинства случаев ОРДС.
Патогенез. ОРДС характеризуется ги-
пергидратацией легочной ткани, но в отличие от кардиогенного отека легких, повышение
гидростатического давления в капиллярах легких не является первичным. Основным
патогенетическим механизмом развития отека легких при ОРДС является патологическое
увеличение |
проницаемости |
альвеолярно- |
капиллярной мембраны. Последнее является |
||
следствием |
непосредственного воздействия |
|
патогенных факторов (токсичных газов, |
||
аспирации |
содержимого |
желудка) или |
Рис. 21.7. Основные причины отека легких. *Самая частая причина. |
|
|
опосредованного влияния активированных
форменных элементов крови, в основном нейтрофилов (последние имеют ключевое значение при сепсисе и/или эндотоксемии). А при вирусных и аутоиммунных процессах ключевое значение имеют лимфоциты.
Повреждение легких при ОРДС вызвано дисбалансом воспалительных и противовоспалительных медиаторов. В качестве вероятного механизма, смещающего баланс в сторону воспалительных цитокинов,
рассматривается гиперактивация транскрипционного фактора NF-kB в алвеолоцитах, эндотелиоцитах и в макрофагах. Как уже известно из раздела «Воспаление», синтез основных провоспалительных цитокинов происходит под действием именно этого фактора транскрипции. Уже через 30 минут после острого воздействия усиливается синтез ИЛ-8 (мощный хемотактический и активирующий нейтрофилы фактор), ИЛ-
1 и ФНО (активируют эндотелий), а также секвестрация нейтрофилов из кровеного русла и дальнейшая активация в капиллярах легких. Нейтрофилы накапливаются в легких по двум механизмам:
1. Накопление нейтрофилов в легких обусловлено большой площадью поверхности легких и выраженной экспрессией молекул адгезии на поверхности эндотелиоцитов и альвеолярных клеток. Увеличивается адгезивная «готовность» нейтрофилов под действием ИЛ-8 и ФНО, они экспрессируют молекулы адгезии (L-
селектины, затем увеличивается сродство интегринов) и их рецепторы, с помощью которых связываются с лигандами активированных эндотелиальных клеток. Эндотелиоциты, активированные цитокинами, и прежде всего ФНО-α и ИЛ-1β, на своей поверхности экспрессируют адгезивные молекулы, в частности ICAM-1.
Таким образом, нейтрофилы «хотят адгезироваться, а эндотелиоциты – не возражают».
2. Активированные нейтрофилы более «жесткие» и менее деформируемые, вследствие этого они остаются в узкой капиллярной сети легких. Фактически, легкие являются «фильтром» для активированных нейтрофилов.
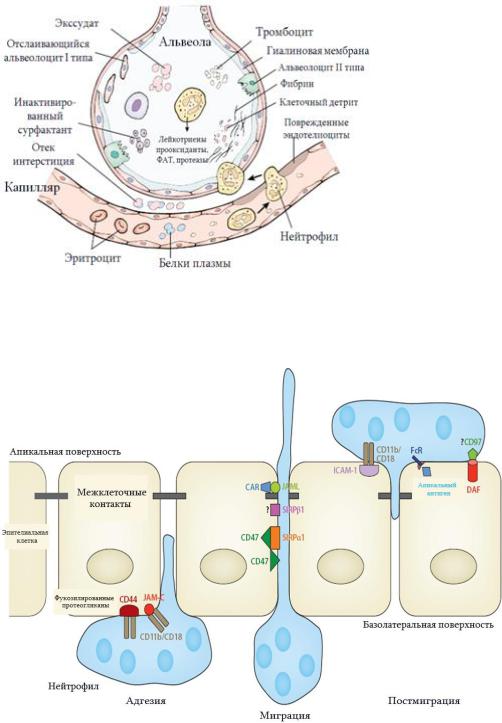
За вышеупомянутым следует миграция лейкоцитов с экзоцитозом и дыхательным взрывом.
Активированные нейтрофилы высвобождают большое количество свободных радикалов, лизосомальных ферментов, ФАТ, лейкотриенов, других производных фосфолипидов, нити хроматина (вследствие активации нетоза), которые повреждают альвеолярно-капиллярную мембрану, альвеолярные клетки и инактивируют сурфактант4 (рис. 21.8). Сочетанное повреждение эндотелия и эпителия повышает проницаемость стенки
4 Сурфактат, покрывая альвеолярную поверхность, уменьшает внутриальвеолярное поверхностное натяжение. Эта система, уменьшая поверхностное натяжение, повышает эластичность легких. Снижение поверхностного натяжения более выражено во время выдоха. Сурфактат обеспечивает равномерное наполнение воздухом альвеол.
13
Кафедра Патофизиологии, Патофизиология системы дыхания, ФОМ, 2021г.
альвеолярных капилляров и делает возможным проникновение жидкости, белков, форменных элементов из крови в интерстициальную ткань легких и в альвеолярную полость. Смесь этих компонентов с фибрином,
образующимся в результате активации каскада коагуляции, формируют гиалиновую мембрану. |
|
|
||||
|
Трансэпителиальная эмиграция нейтрофилов |
|||||
|
осуществляется тремя последовательными стадия- |
|||||
|
ми: адгезии, миграции и постмиграции (рис. 21.9). |
|||||
|
В первой стадии нейтрофил прикрепляется к базо- |
|||||
|
латеральной поверхности альвеолярного эпителия |
|||||
|
посредством интегринов-β2. По видимому, интег- |
|||||
|
рин CD11b/CD18 является первичной молекулой |
|||||
|
адгезии. В процессе миграции важнейшую роль |
|||||
|
играет CD47, который экспрессирован на |
|||||
|
поверхности как нейтрофила, так и альвеолоцита. В |
|||||
|
дальнейшем (в стадии постмиграции) нейтрофилы, |
|||||
|
оставаясь |
прикрепленными |
к |
апикальной |
||
|
поверхности |
альвеолоцитов, |
осуществляют |
|||
Рис. 21.8. Патогенез ОРДС. |
фагоцитоз |
и |
уничтожение |
бактерий. |
В |
|
|
|
|
|
|
|
|
|
физиологических |
условиях |
они |
способствуют |
||
закрыванию образовавшегося после их прохождения отверстия. При ОРДС, однако, эти отверстия столь
многочисленны, что не могут закрыться, что способствует накоплению экссудата в альвеолах. |
|
|
||||
|
|
Миграция |
нейтрофилов |
|||
|
при |
ОРДС |
выраженна |
|||
|
настолько, что часто приводит |
|||||
|
к развитию |
нейтропении в |
||||
|
периферической крови. |
|
||||
|
|
В воспалительный |
про- |
|||
|
цесс вовлекаются также моно- |
|||||
|
нуклеарные |
фагоциты легоч- |
||||
|
ной паренхимы. Их активации |
|||||
|
способствуют гипоксия, дли- |
|||||
|
тельная эндотоксемия и нали- |
|||||
|
чие |
некоторых |
активирован- |
|||
|
ных компонентов комплемента |
|||||
|
(C5a). Последние осуществ- |
|||||
|
ляют |
массивное |
высвобожде- |
|||
|
ние |
провоспалительных |
ме- |
|||
|
диаторов, таких как ФНО-α, |
|||||
Рис. 21.9. Эмиграция нейтрофилов в альвеолы. Примечание: DAF (decay-accelera- |
||||||
|
|
|
|
|
||
ting factor) - известен также как один из регуляторов в системе комплемента, его |
протеолитические ферменты, |
|||||
недостаточность приводит к гемолизу при пароксизмальной ночной гемоглобину- |
АФК, ИЛ-1 и так далее. |
|
||||
рии, JAM-C - компонент десмосом, лиганд CD11b/CD18, по которой осуществ- |
|
При |
вирусных |
и |
||
ляется трансэпителиальная эмиграция нейтрофилов, JAM (Junctional Adhesion |
|
|||||
аутоиммунных |
процессах |
|||||
Molecule) - молекула адгезии межклеточных контактов, SIRP – signal regulatory |
||||||
|
|
|
|
|
||
protein (как и ICAM, VCAM, принадлежит к семейству иммуноглобулинов). |
патогенетическими факторами |
|||||
|
повреждения |
|
альвеоло- |
|||
|
|
|||||
капиллярной мембраны являются лимфоцитарная инфильтрация и иммунные комплексы.
Все вышеупомянутые факторы увеличивают проницаемость микрососудов легких, обуславливая развитие отека и разрушение альвеолярной мебраны. Альвеолы заполняются воспалительным экссудатом и
14

Кафедра Патофизиологии, Патофизиология дыхания, ФОМ, 2021г.
прогрессируют рестриктивные нарушения вентиляции. По сути, возникает и развивается «лишенное биологической мотивации (смысла) гипервоспаление». В этот процесс вовлекаются не только легкие, но и другие внутренние органы, способствуя развитию полиорганной недостаточности.
Активация и повреждение эндотелиоцитов становится причиной образования микротромбозов в легких,
которые к диффузному альвеолярному повреждению добавляют ишемическое поражение легочной паренхимы. Для ОРДС характерны местные нарушения гемостаза в легких. В сыворотке крови и бронхоальвеолярном лаваже5 уровень тканевого фактора повышается, а антикоагулянтов - снижается.
Коагуляционный каскад является мощным провоспалительным сигналом6. К примеру, тромбин способствует адгезии нейтрофилов на эндотелии.
Все перечисленные процессы усугубляют альтерацию и в итоге повреждают альвеолярные клетки II
типа, которые синтезируют сурфактант (рис. 21.8). Следует отметить, что до этого инактивирующий или повреждающий эффект на систему сурфактанта оказывали проникшие в альвеолы плазменные белки
(например, фибриноген) и повреждающий фактор. Заметим, что повреждение сурфактанта и угнетение его синтеза вызывают и медиаторы воспаления (как непосредственно, так и путем ишемии легких). В итоге развиваются множественные микроателектазы, усугубляются рестриктивные нарушения вентиляции и диффузии, гипоксемия, развивается внутрилегочное шунтирование крови (из-за чего гипоксемия не коррегируется оксигенотерапией).
Таким образом, патогенетическими факторами развития ОРДС являются:
1. гиповентиляция, в основе развития которой лежат рестриктивные нарушения, а обструктивные
(бронхоспазм, развивающийся под действием различных БАВ) могут иметь определенное значение в усугублении состояния,
2.нарушения диффузии,
3.нарушения перфузии легких. Медиаторы вызывают спазм сосудов легких, развивается легочная гипертензия, усиливается тромбообразование, вызывающее легочную гипертензию и внутрилегочное шунтирование крови.
Вслучае благоприятного исхода ОРДС экссудат рассасывается7, мертвые клетки удаляются и заменяются новыми эндотелиальными и эпителиальными клетками. Удаление экссудата и тканевого детрита осуществляют макрофаги (как и в остальных случаях повреждения). Эпителиальные клетки (альвеолоциты I
типа) восстанавливаются за счет пролиферации сохранившихся альвеолоцитов II типа, которые покрывают обнаженную базальную мембрану. Было выявлено, что в процессе восстановления участвуют стволовые клетки бронхо-альвеолярного эпителия. Восстановление эндотелия происходит за счет миграции эндотелиоцитов из неповрежденных капилляров, а также эндотелиальных клеток-предшественниц.
При неблагоприятном течении ОРДС примерно через 7 дней начинает развиваться фиброз легких.
Снижается функциональный остаточный объем и усугубляются патологическое внутрилегочное шунтирование крови и дыхательная недостаточность. Если этот синдром не подвергнуть обратному развитию,
то ОРДС приведет к смерти.
Респираторный дистресс синдром новорожденных (болезнь гиалиновых мембран). Респираторный дистресс синдром новорожденных чаще развивается у недоношенных новорожденных, т.к. альвеолярные клетки II типа, синтезирующие сурфактант формируются к 25-28 неделям внутриутробной жизни, а их полное «созревание» происходит к 38 недели беременности. Синтез сурфактанта активируется кортизолом и подавляется инсулином. Незрелость дыхательной системы и недостаточность сурфактанта у недоношенных
5Жидкость, образуемая при обмывании бронхиального дерева и при обратном ее вытягивании.
6Мы рассматривали основных участников этого каскада как плазменные медиаторы воспаления.
7Основным механизмом удаления является смещение воды за натрием, чему способствует экспрессируемая на базолатеральной поверхности эпителиальных клеток Na-K-вая АТФ-аза. Жидкость, удаленная из альвеол, впоследствии по лимфатическим путям, плеврой удаляется из легочной ткани.
15
Кафедра Патофизиологии, Патофизиология системы дыхания, ФОМ, 2021г.
детей приводит к развитию альвеолярного коллапса. Частота этого синдрома повышена у новорожденных мужского пола, белой рассы, у детей, рожденных кесаревым сечением до 38 недель беременности, у детей рожениц с сахарным диабетом, детей у которых была асфиксия или гипотермия.
Удетей, матери которых страдают СД, вероятность развития ОРДС выше почти в 6 раз. Вследствие
гипергликемии матери страдающей диабетом, в организм плода через плаценту проникает большое количество глюкозы, вследствие чего у плода развивается гипертрофия -клеток поджелудочной железы и
гиперинсулинемия. А последняя угнетает синтез сурфактанта.
Уноворожденных, родившихся с помощью кесарева сечения, высокая вероятность развития этого синдрома вероятно обусловлена отсутствием стресса, развивающегося при прохождении по родовым путям,
вследствие которого повышается уровень кортизола. Принимая во внимание данные наблюдения, перед родами таким роженицам назначают кортикостероиды.
При рождении во время первого вдоха для расправления легких необходимо высокое давление. При нормальном уровне сурфактанта после дыхательного акта в легких удерживается до 40% остаточного объема,
и для дальнейшего дыхания требуется меньшее давление. При недостаточности сурфактанта между дыхательными актами легкие закрываются, заставляя новорожденного совершать при каждом новом вдохе ту же работу, что и при первом. Безвоздушный отдел легких становится твердым и плохо расправляющимися
(растягивающимся). Внутри альвеол формируются гиалиновые мембраны, вследствие инфильтрации богатой белками и фибрином жидкости в альвеолярное пространство. Фибрино-гиалиновые мембраны создают барьер для газообмена, приводя к развитию гипоксемии. Это состояние еще больше нарушает синтез сурфактанта.
Основная разница респираторного дистресс-синдрома взрослых и новорожденных заключается в том,
что у взрослых нормальное количество сурфактанта инактивируется, разрушается (вторичная недостаточность), а у новорожденных изначально существует дефицит сурфактанта (первичная недостаточность).
16