

книги / Электрохимическая коррозия и защита металлов
..pdfи производимый ею эффект зависят от потенциала. Это, в частности, может приводить к отклонению кривой Е—lgi, характеризующей электрохимическое поведение металла, от обычной прямолинейной зависимости, что наблюдается, например, при растворении железа в солянокислых растворах. Стимулирующая адсорбция — процесс очень быстрый по сравнению с адсорбцией ингибирующей.
3.4.Растворение железа в кислых растворах. Механизм растворения Дж. Бокриса
Стандартный электродный потенциал железа EFe0 2 + / Fe = −0, 44 В,
а EFe0 3+ / Fe = – 0,037 В. Более отрицательное значение стандартного
потенциала при растворении железа диктует образование двухзарядных катионов Fe2+. Рассмотрим механизм растворения, предложенный Дж. Бокрисом и получивший широкое распространение. Этот механизм не учитывает участия анионов раствора в процессе растворения и поэтому требует внесения существенных поправок, что будет показано ниже.
Анодное растворение железа в кислых сульфатных растворах ускоряется с ростом рН раствора. Тафелевы прямые для растворов
спостоянной концентрацией сульфата, но переменной кислотностью
сростом рН закономерно смещаются в направлении, соответствующем сдвигу потенциала в отрицательную сторону. Наклон тафелевых прямых, независимо от рН, составляет 40 мВ (рис. 6). Эффект ускорения растворения характеризуется десятикратным увеличением тока (при Е = const) при увеличении рН на единицу (рис. 7). Другими словами, порядок реакции растворения железа по ионам водорода равен –1. Это с учетом найденного тафелевого наклона дает возможность принять следующую схему процесса. На поверхности железа
адсорбируются молекулы Н2О, подвергаясь при этом диссоциации. Такая диссоциативная адсорбция приводит к образованию поверхно-
стного адсорбированного комплекса Fe(OH)адс− :
Fe + H2O ←→ K1 |
Fe(OH)адс− + H+. |
(3.36) |
|
|
41 |

За адсорбционной стадией (3.36) следует электрохимическая реакция отщепления первого электрона от атома железа, связанного с частицей ОН–:
Fe(OH)− |
↔ Fe(OH) |
адс |
+ е. |
(3.37) |
адс |
|
|
|
Рис.6. Анодные поляризационные кривые |
Рис. 7. Зависимость скорости растворения |
для железа в растворах H2SO4 +Na2SO4 при |
железа в растворах H2SO4 +Na2SO4 |
С(SO2–4 ) = 0,5 моль/л и различных рН: |
при Е= – 0,2 В от рН |
1 — 0,2; 2 — 1,2; 3 — 2,3 |
при С(SO24− ) = 0,5 моль/л |
Стадия (3.37) предшествует замедленной стадии отщепления второго электрона и поэтому является равновесной. Стадия, лимитирующая скорость всего процесса, имеет вид
Fe(OH) |
адс |
→ Fe(OH) + + e. |
(3.38) |
|
|
|
Она сопровождается диссоциацией комплекса Fe(OH)+ в растворе:
Fe(OH) + + Н+ ↔ Fe2+ + H2O. (3.39)
С учетом замедленности стадии (3.38) для скорости растворения железа справедливо уравнение:
ia = KaFe(OH)адс exp(β FE / RT ). |
(3.40) |
Величину aFe(OH )адс можно найти из уравнения Нернста для ста-
дии (3.37):
42
|
0 |
|
RT |
aFe(OH)адс |
|
|
|
(3.41) |
|||
|
E = E(3.37) + |
|
|
|
|
|
|
|
|
||
|
|
F ln а |
– . |
|
|
||||||
|
|
|
|
|
|
||||||
|
|
|
|
|
|
Fe(OH) |
|
|
|
|
|
|
|
|
|
|
|
адс |
|
|
|
|
|
Для этого следует подставить в него aFe(OH )− |
из уравнения дис- |
||||||||||
|
|
|
|
|
|
|
|
адс |
|
|
|
социации для стадии (3.36): |
|
|
|
|
|
|
|
|
|
|
|
|
aFe(OH )− |
|
= K1 / aH+ . |
|
|
|
(3.42) |
||||
|
адс |
|
|
|
|
|
|
|
|
|
|
Совместное решение уравнений (3.41) и (3.42) приводит к вы- |
|||||||||||
ражению |
|
|
|
|
|
|
|
|
|
|
|
|
aFe(OH )адс = K / aH−1+ exp(FE / RT ), |
|
(3.43) |
||||||||
где K / — новая константа, объединяющая константу диссоциации |
|||||||||||
K1 (уравнение (3.36)) и постоянную величину |
E0 |
|
F |
||||||||
|
(3.37) |
|
. |
||||||||
|
RT |
|
|||||||||
|
|
|
|
|
|
|
|
|
|
||
Подстановка (3.43) в (3.40) дает уравнение для скорости рас- |
|||||||||||
творения железа: |
|
(1 + β |
) FE / RT . |
|
|
|
|||||
|
ia = K / a−1+ exp |
|
(3.44) |
||||||||
|
H |
|
|
|
|
|
наклон ∂Е/∂lgia = |
||||
Требуемые |
уравнением (3.44) |
тафелев |
|
||||||||
= 2,3RT/(1+β)F (при β = 0,5 равный 40 мВ) и порядок реакции по |
|||||||||||
ионам Н+ ∂ lg ia ∂ |
lg aH+= − 1 при Е = const действительно совпадают |
||||||||||
сэкспериментальными значениями.
Растворение этого металла ускоряется не только с ростом рН, но (при постоянных рН) и
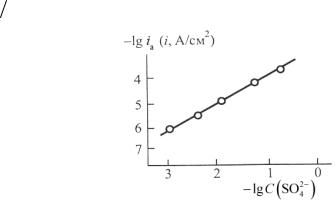
сростом концентрации анионов раствора, например сульфата (рис. 8), причем порядок реакции по анионам близок к единице.
Такой результат согласу- |
Рис. 8. Зависимость скорости растворения |
|
ется с предположением о том, |
железа в растворах H2SO4 +Na2SO4 |
|
при Е = – 0,2 В от C(SO24− ) при рН = 3 |
||
что за рассмотренными выше |
||
|
||
|
43 |
стадиями (3.36) и (3.37) следуют стадии: |
|
|
|
Fe(OH)адс + (HSO4 )адс– |
= Fe2+ + SO42− |
+ H2O + e |
(3.45) |
или |
|
|
|
Fe(OH)адс + (SO42− )адс |
= Fe2+ + SO42− |
+ ОН– + е. |
(3.46) |
Они лимитируют скорость всего процесса. При этом совокупности последовательных реакций (3.36), (3.37), (3.45) и (3.36), (3.37), (3.46) протекают параллельно, так что для суммарной скорости растворения железа можно записать:
ia = K / aFe(OH) |
a |
|
− exp(β FE / RT ) + |
|
|
адс |
HSO4 |
|
|
+K / / aFe(OH) |
a |
2 |
− exp(β FE / RT ), |
(3.47) |
|
адс |
SO4 |
|
|
где K / , K / / — константы равновесия реакций (3.45) и (3.46) соответственно.
Если принять, что константы скоростей реакции с участием ионов обоих видов близки по величине (K/ ≈ K// = K), то уравнение (3.47) можно записать в виде
i = Ka |
|
(a |
− + a 2 |
− ) exp(β FE / RT ) , |
(3.48) |
|
a |
Fe(OH)адс |
HSO4 |
SO4 |
|
|
|
или, обозначая сумму |
(aHSO4− + aSO42− ) |
через ас, а также учитывая |
||||
(3.36) и (3.37) и логарифмируя, в виде |
|
|
||||
lg ia = K/// + pH + lg ac + (1 + β) FE/2,3RT, |
(3.49) |
|||||
где K // / = lg K / K / / . |
|
|
|
|
|
|
С последним уравнением хорошо согласуется найденная экспериментально зависимость скорости растворения железа от состава кислых сульфатных растворов, показавшая, что lg ia при E = const линейно связан с логарифмом суммарной концентра-
ции сульфата C (SO24− ) при постоянном рН (см. рис. 8). Лога-
рифм плотности тока линейно связан с рН при постоянной концентрации сульфата и при постоянном значении потенциала
(см. рис. 7).
44
3.5. Механизм растворения К.Е. Хойслера
При исследовании растворения железа К.Е. Хойслером были получены данные, отличные от полученных Дж. Бокрисом и другими: тафелев наклон ba = 30 мВ, порядок реакции по ОН–-ионам равен 2. Это различие Хойслер объяснял тем, что механизм растворения, названный его именем, выполняется в стационарных условиях, т.е. при длительных выдержках при каждом потенциале и достижении стационарного значения плотности тока. Процесс протекает в четыре стадии:
Fe + OH– ↔ Fe(OH)адс + е, |
|
(3.50) |
||
Fe(OH)адс + Fe ←→ |
K2 FeFe(OH)адс, |
|
(3.51) |
|
– |
K |
Fe(OH)адс + FeOH |
+ |
+ 2e, (3.52) |
FeFe(OH)адс + ОН |
1 → |
|
||
FeOH+ + H+ ↔ Fe2+ + H2O, |
|
(3.53) |
||
где FeFe(OH)адс или Fe(OH)адс — поверхностный комплекс, автокаталитически ускоряющий растворение железа.
При большом времени экспозиции образцов при каждом потенциале устанавливается адсорбционное равновесие на стадиях
(3.50) и (3.51). Стадии (3.50), (3.51), (3.53) протекают быстро,
быстро достигают равновесия (равновесные стадии), стадия (3.52) медленная, лимитирующая, определяет скорость всего процесса. Скорость этой двухэлектронной стадии определяется выражением:
i |
= K |
FeFe(OH) |
|
[OH− ]exp |
|
2β FE |
. |
(3.54) |
|
|
|||||||
a |
1 |
|
адс |
|
|
|
|
|
|
|
|
|
|
|
RT |
|
|
Но концентрация [FeFe(OH)адс] равна концентрации [Fe(OH)адс]. Последнюю находим по уравнению Нернста для равновесия (3.50):
E = E0 + |
RT |
ln |
[Fe(OH)адс] |
. |
|
(3.55) |
||
|
F |
[OH− ] |
|
|
|
|
||
Из уравнения (3.55) находим концентрацию |
|
|
||||||
|
|
|
|
(E − E |
0 |
)F |
|
|
[Fe(OH)адс] = [OH− ]exp |
|
. |
(3.56) |
|||||
|
|
|
||||||
|
|
|
|
RT |
|
|
|
|
|
|
|
|
|
|
|
|
45 |
|
|
|
E0 F |
|
|
|
|
||||
Вводя константу K = exp − |
|
|
|
, подставляя (3.56) в (3.54) |
|||||||
|
|
|
|||||||||
|
|
|
RT |
|
|
|
|||||
и объединяя константы K1 (см. уравнение (3.54)) и K в K1/ , |
находим: |
||||||||||
/ |
− 2 |
(1 + 2β )FE |
|
||||||||
ia = K1 [OH |
] exp |
|
|
|
|
. |
(3.57) |
||||
|
|
|
|
||||||||
|
|
|
|
|
|
RT |
|
|
|
||
Логарифмируя выражение (3.57) и переходя к десятичным ло- |
|||||||||||
гарифмам, находим выражение для потенциала: |
|
||||||||||
E = a − |
2,3 2RT |
pH + |
|
2,3RT |
|
lg ia . |
(3.58) |
||||
|
|
F (1+ 2β |
) |
||||||||
|
F (1 + 2β ) |
|
|
|
|
|
|
||||
Тафелев наклон b = ∂ E /∂ |
lg ia= 30 мВ при постоянном рН. По- |
||||||||||
рядок по ОН–-ионам равен 2, или (∂ lg ia /∂ pH )E =const= − 2 при постоянном потенциале.
3.6. Влияние анионов на кинетику растворения металлов
Влияние анионов на скорость растворения впервые обнаружил М.Н. Центнершвер на примере растворения алюминия. При практически одинаковой концентрации ионов водорода (~1 н.) скорость растворения Al в серной кислоте была в 40 раз ниже, чем в соляной кислоте. Весьма медленное растворение алюминия в растворе серной кислоты сильно ускоряется анионами хлора, введенными в виде солей калия (KCl). Значительно меньше влияет на скорость растворения ион брома, и почти не активирует растворение ион йода.
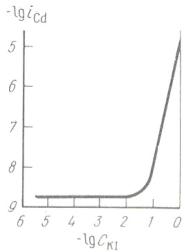
Другие анионы также ускоряют процесс растворения металлов. Убедительный пример ускоряющего влияния концентрации иодида калия на скорость растворения кадмия показан на рис. 9.
Начиная с некоторой «критической» концентрации соли, в данном случае 0,1 н., дальнейшее повышение содержания этой соли в 10 раз сопровождается увеличением скорости анодного процесса почти в 1000 раз.
46
Согласно А.Я. Шаталову экспериментальные результаты по ускоряющему действию анионов на ионизацию металлов можно объяснить, лишь предположив, что элементарная стадия процесса ионизации происходит при участии анионов, например, по уравнению
Me + mAn |
− |
→ MeAn |
m−n |
|
|
|
+ ne. (3.59) |
|
|
||
При этом образуется поверхност- |
|
|
|||
ный комплекс с частичным перено- |
|
|
|||
сом заряда. |
|
|
|
|
|
В этом случае скорость иони- |
|
|
|||
зации металла будет включать кон- |
Рис. 9. Зависимость скорости |
||||
центрацию анионов в степени m и |
растворения кадмия iCd |
|
|||
уравнение, описывающее этот про- |
от концентрации KI в растворе |
||||
цесс, примет вид |
|
|
|
||
|
|
|
i = K[An− ]m exp(β FE / RT ). |
(3.60) |
|
Показатель степени m в этом уравнении зависит от природы металла и от вида аниона.
В большинстве случаев стимулирующее действие анионов объясняют специфической адсорбцией этих ионов на поверхности металлов. Эта специфическая адсорбция анионов начинается при потенциалах, намного меньших, чем потенциал коррозии. Ее следует рассматривать как начало образования соответствующего соединения на поверхности металла (за счет ковалентных связей). Прочность этой связи ниже, чем в индивидуальном соединении. При смещении потенциала в положительную сторону связь галоидных комплексов с поверхностью металла уменьшается, и они переходят в раствор (акт растворения). Механизм растворения можно представить в виде двух последовательных реакций:
Me + mCl− = (MeClm )m− , |
(3.61) |
(MeClm )m− → MeCl(mn−m)+ ne. |
(3.62) |
|
47 |
Первая из них соответствует специфической адсорбции аниона на поверхности металла с образованием поверхностного комплекса, а вторая — ионизации металлического атома, входящего в этот комплекс.
Стимулирующее влияние данного аниона на процесс анодного растворения может проявляться только при достижении определенного критического потенциала, при котором прочность связи поверхностного атома металла с адсорбированным ионом становится равной прочности ковалентной связи в соответствующем индивидуальном соединении. Разным анионам должны соответствовать разные значения критического потенциала. Таким образом, если при данном потенциале на поверхности металла адсорбируются одновременно несколько компонентов раствора, то переходить в раствор могут не все из образовавшихся поверхностных комплексов, а только те, для которых прочность химической связи достигла определенной величины, причем при условии, что связь этого комплекса с раствором прочнее связи комплекса с металлической поверхностью.
3.7. Ингибирующее действие анионов
Если связь поверхностного комплекса [Mem Ann ]адсm−n с металлом
будет прочнее, чем с раствором, то происходит блокировка элементарных актов растворения металла, т.е. замедление, или ингибирование, коррозии. Ингибирующее действие могут оказывать галидионы в некоторых случаях. Таким образом, роль галид-ионов двойственна: они могут стимулировать, т.е. ускорять коррозию, а могут, наоборот, ингибировать, т.е. замедлять ее.
Известно, например, что ионы I– тормозят ионизацию никеля в кислых растворах сульфат-ионов. Этот эффект может быть понят как результат конкурирующей адсорбции между ионами йода и гидроксила, если предположить, что первичная неэлектрохи-
48
мическая стадия ионизации никеля в сульфатном растворе состоит в образовании поверхностных комплексов металла с OH–- ионами:
Ni + mH2O → |
m− |
+ |
. |
Ni(OH)m+ |
mH |
Никель в этом случае будет растворяться в результате последующего превращения поверхностного комплекса:
Ni(OH)mm− → Ni(OH)(mm+n )+− ne.
Как показывают расчеты, скорость анодного растворения никеля при этом пропорциональна концентрации OH–-ионов в степени 1,5, т.е. она возрастает быстрее, чем по линейному закону.
Гидроксильные ионы в присутствии ионов йода частично будут замещены на поверхности никеля I–-ионами, что в итоге приводит
кснижению стимулирующего действия OH–-ионов, т.е. по сути дела
кингибированию всего процесса.
Те же самые представления позволяют объяснить ингибирующее действие добавок галидных ионов на растворение железа в кислом сульфатном растворе, а также ряд других случаев.
3.8. Зависимость стационарной скорости ионизации металла от потенциала
Так как коррозионные разрушения металла обычно непосредственно вызываются реакцией ионизации, то знание особенностей этой реакции имеет важное значение для решения коррозионных проблем. В первую очередь, это относится к закономерностям стационарной ионизации, поскольку в большинстве случаев при достаточно длительном контакте металла с агрессивной средой коррозионный процесс стабилизируется. Важнейшей характеристикой коррозионного поведения металла в подобных условиях является зависимость стационарной скорости ионизации (скорости растворения) iст от потенциала Е.
49

3.8.1. Общая характеристика зависимости iст от потенциала Е
Типичная кривая iст = f (E) представлена на рис. 10 и показы-
вает главные области растворения по различным механизмам. Точка А отвечает бестоковому значению потенциала Eкор. Ток коррозии iкор можно рассчитать по значению Km− (по формуле (1.2)).
Тафелевский участок кривой АВ — область активного растворения (активная область). Скорость растворения iст здесь растет с увеличением потенциала в связи с действием ускорения электрохимической реакции по тафелевой зависимости.
При потенциалах положительнее т. В становится заметным торможение процесса, связанное с началом пассивации
металла. Пассивация обусловлена образованием защитного слоя, который при дальнейшем повышении потенциала (участок ВС, так называемая переходная область, падающий участок) продолжает распространяться по поверхности электрода. Потенциал максимума тока, который для достаточного развития пассивации должен быть обязательно превышен, часто называют критическим потенциалом пассивации (Екр), а соответствующую плотность тока — критическим током пассивации (iкр).
Область СD, CF, в которой стационарная скорость растворения iст невелика (10–5–10–8 А/см2) и практически не зависит от потенциала, называют областью пассивного состояния или пассивной областью (здесь iст = iп, где iп — плотность тока в пассивной области). Рост скорости ионизации при увеличении потенциала здесь полно-
50