

СМА биотех
.docxСпинальная атрофия является нервно-мышечным заболеванием. У детей с этой патологией наблюдаются различной степени выраженности атрофия мышечной ткани с преимущественным вовлечением проксимальных групп мышц и поражение нижних конечностей в начале заболевания. Клиническая картина СМА в целом включает в себя разнообразные проявления, но всегда сопровождается избирательной дегенерацией двигательных нейронов спинного мозга в сочетании со сложным комплексом сопутствующих проявлений, что указывает на важную системную роль белка SMN (белок выживаемости мотонейронов, он поддерживает стабильность и полноценное функционирование мотонейронов).
К клиническим проявлениям СМА относятся:
• бульбарные нарушения: ослабленный крик у детей, фасцикуляции языка;
• нарушения дыхания;
• нарушения вегетативной регуляции сердца и врожденные пороки сердца;
• гастроэзофагеальный рефлюкс;
• запор, потеря массы тела;
• контрактуры суставов;
• снижение или отсутствие глубоких рефлексов;
• атрофия мышц, их слабость и гипотония;
• у ряда пациентов может наблюдаться грубый постуральный тремор – мини-полимиоклонус.
Традиционно СМА разделяется на 5 подгрупп
по убыванию тяжести заболевания с учетом
возраста дебюта болезни, потенциальных
возможностей двигательного развития
и типичного возраста смерти. В табл
представлена общая характеристика СМА,
обусловленных мутацией в гене SMN1
(расположен на 5 хромосоме в локусе
5q13).

Причины. КРАТКО СМА вызвана мутацией в гене SMN1, который в норме производит белок SMN. Из-за мутации гена у людей с СМА производится меньшее количество белка SMN, что приводит к потере моторных нейронов.
ПОДРОБНО. Генетической причиной СМА являются мутации в гене SMN1. Ген SMN1 кодирует белок, необходимый для работы двигательных нейронов. Важной частью этого белка является фрагмент, информация о котором содержится в 7 экзоне (определенной части) гена SMN1. Любая мутация, которая приводит к отсутствию этого фрагмента в белке или же значительно нарушает строение белка, может привести к развитию заболевания. Полное отсутствие 7 экзона (делеция) является самой частой причиной СМА. Если на обеих хромосомах в гене SMN1 есть нарушения в 7 экзоне, то нормальный белок не будет формироваться, а отсутствие нормально функционирующего белка вызывает СМА.
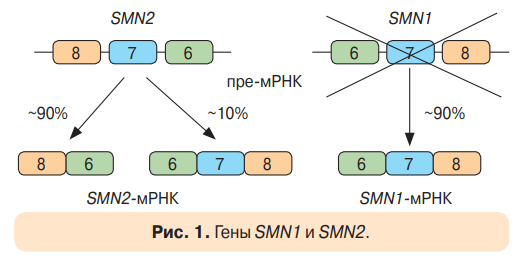
В геноме человека также есть ген SMN2.
Гены SMN1 (теломерная копия) и SMN2 (центромерная
копия) отличаются по кодирующей
последовательности только одним
нуклеотидом. Эта нуклеотидная замена
в 7-м экзоне гена SMN2 приводит к изменению
сплайсинга РНК и отсутствию 7-го экзона
примерно в 90% транскриптов гена SMN2 (рис).
По этой причине ген SMN2 является источником
синтеза измененной, нестабильной и
быстро разрушающейся изоформы белка
SMNΔ7, неспособного компенсировать
последствия делеций в SMN1. Однако, в
результате ошибок синтеза белка, которые
иногда случаются в нашем организме, с
гена SMN2 может синтезироваться небольшое
количество правильного, функционального
белка, необходимого для работы двигательных
нейронов. Именно поэтому даже при полном
отсутствии нормального белка с гена
SMN1 количество копий гена SMN2 влияет на
тяжесть симптомов.
Стандартная терапия. Этиологического лечения спинальных мышечных атрофий на сегодняшний день не существует. Больные СМА нуждаются в специальном диетическом питании, поддерживающей терапии и многих других попечительских действиях, иначе отмечается усиление отрицательной динамики.
Большинство традиционно применяемых препаратов (перечислены ниже) обладают общим неспецифическим механизмом действия и их применение имеет невысокий уровень доказательности.
L-Карнитин – аминокислота, природное вещество, родственное витаминам группы В. В отличие от витаминов, карнитин синтезируется в организме, поэтому его называют витаминоподобным веществом. В организме человека присутствует в тканях поперечнополосатых мышц и печени. Является фактором метаболических процессов, обеспечивающих поддержание активности кофермента А (КоА).
Коэнзим Q10 (кофермент Q10, убихинон) — это группа коферментов — бензохинонов, содержащих хиноидную группу (отсюда обозначение Q) и содержащих несколько изопрениловых групп (например, 10 в случае кофермента Q10). Убихиноны — это жирорастворимые коферменты, представленные преимущественно в митохондриях эукариотических клеток. Убихинон является компонентом цепи переноса электронов и принимает участие в окислительном фосфорилировании.
Церебролизин – ноотропное средство, комплексный препарат с нейротрофической активностью, который используется для лечения больных с различными неврологическими заболеваниями, такими как ишемический и геморрагический инсульты, сосудистая деменция, включая болезнь Альцгеймера.
Актовегин – препарат, содержащий низкомолекулярные пептиды и производные аминокислот. Актовегин, будучи гемодериватом, который получают посредством диализа и ультрафильтрации (проходят соединения с молекулярной массой менее 5000 дальтон), содержит только физиологические вещества. На молекулярном уровне Актовегин вызывает увеличение утилизации и потребления кислорода (повышает устойчивость к гипоксии), повышает энергетический метаболизм и потребление глюкозы.
Итд (Солкосерил, Витамины В1, В6, В12 (Нейромультивит), Витамин Е, Вальпроаты – оказывают миорелаксирующее, седативное действие, проявляют противоэпилептическую активность при всех типах эпилепсий, Сальбутамол, Мотилиум) Все поддерживающая терапия итп.
Таргетная терапия. Каждый наверняка слышал о самом дорогом препарате на Земле (2,125 млн долларов). Золгенсма, или же Онасемноген абепарвовек. Он является препаратом генной терапии, разработанным для лечения больных проксимальной спинальной мышечной атрофией, вызванной нарушением в гене SMN1. AVXS-101 доставляет синтетическую функциональную копию гена SMN1 в клетки двигательных нейронов, используя аденоассоциированный вирус-вектор 9 серотипа (AAV9). Онасемноген абепарвовек представляет собой генно-терапевтическое лечение, обеспечивающее замену отсутствующего или дефектного гена SMN1 на его функциональную копию, что приводит к нормализации выработки белка выживаемости мотонейронов (SMN). Полноценный ген SMN1 находится внутри вектора аденоассоциированного вируса (adeno-associative virus 9, или AAV9). Это представитель семейства парвовирусов (семейство самых мелких ДНК-содержащих сферических вирусов, лишенных липопротеидной оболочки), который способен инфицировать клетки человека и других приматов, но при этом не является патогенным. Собственный генетический материал вируса удалён и заменён на функционально полноценный ген SMN1. После того, как ген прибывает в нужную локацию, вектор разрушается и выводится из организма. Благодаря способности онасемногена абепарвовека преодолевать гематоэнцефалический барьер (ГЭБ), с последующей трансдукцией (процесс переноса ДНК между клетками при помощи вирусов) клеток-мишеней, подтверждена экспрессия SMN в мотонейронах во всех отделах головного и спинного мозга.
Клинически доказано, что однократная инфузия препарата способна восстановить SMN-экспрессию в моторных нейронах, лишённых функционального гена SMN1